Elena Sanmartín1, 2 ; Adela Cañete2, 3; Victoria Castel2, 3; Jaime Font de Mora1, 2
1 Laboratorio de Biología Molecular y Celular, Instituto de Investigación Sanitaria La Fe, Valencia, España
2 Grupo de Investigación Clínica y Traslacional en Cáncer, Instituto de Investigación Sanitaria La Fe, Valencia, España
3 Unidad de Oncología Pediátrica, Hospital Universitario y Politécnico La Fe, Valencia, España
El neuroblastoma es el tumor extracraneal sólido más frecuente en la infancia y es, después de los accidentes domésticos, la segunda causa más frecuente de mortalidad en niños. Cerca de 700 nuevos casos son diagnosticados cada año en Estados Unidos y 900 nuevos casos en la Unión Europea (EU28). La prevalencia es de 1 caso por cada 7.000 nacimientos vivos. El neuroblastoma es de origen neuroectodérmico y deriva de células embrionarias de la cresta neural. Aunque globalmente la supervivencia del neuroblastoma está por encima del 70%, existen diferentes subtipos con un amplio espectro de comportamiento. Por un lado, los lactantes menores de 18 meses tienen mejor pronóstico, incluso algunos casos experimentan regresión completa de su enfermedad, aún con enfermedad metastásica. Sin embargo, los pacientes mayores de 5 años presentan con mayor frecuencia enfermedad metastásica con mal pronóstico a pesar de recibir tratamientos muy agresivos. La estratificación de los pacientes por grupos de riesgo permite adecuar el tratamiento a partir de factores pronósticos como la edad, la clasificación histopatológica, el estadio y factores de biología molecular como la amplificación MYCN, la deleción 1p y la ploidía. Sin embargo, las alteraciones genéticas descritas hasta el momento no explican el comportamiento tan desigual de este tumor en los diferentes grupos de riesgo. Por ello, la identificación de biomarcadores genéticos implicados en la patogénesis del neuroblastoma, sería de vital importancia para optimizar la clasificación en grupos de riesgo, evaluar adecuadamente la respuesta al tratamiento y definir nuevos enfoques terapéuticos que permitan mejorar las expectativas de curación de los pacientes.
Con el objetivo de identificar nuevas dianas terapéuticas y predictores de respuesta en neuroblastoma, nos planteamos analizar una cohorte de 106 tumores primarios mediante secuenciación masiva. Para ello diseñamos e implementamos un panel de genes personalizado que contenía 483 amplicones de 26 genes con implicación en neuroblastoma u otros cánceres. El análisis de secuenciación masiva se llevó a cabo en la Unidad Genómica del Instituto de Investigación Sanitaria La Fe con los sistemas Ion PGM/Ion Proton (Life Technologies). En el análisis de los datos de secuenciación masiva se filtraron sólo las variantes alélicas de baja frecuencia (MAF <1% en 1000 G) que se localizan en exones o sitios de splicing. Para investigar el potencial patogénico de las variantes detectadas, se estudiaron las bases de datos de variantes (COSMIC, dbSNP, ClinVar), los algoritmos de predicción de función proteica (SIFT, PolyPhen-2 or Mutation Taster) o las herramientas de predicción de efecto de splicing (SpliceView, NetGene2, Human Splicing Finder, NNSplice).
Los resultados de este estudio publicado en Oncotarget mostraron que 62 de los 106 casos de neuroblastoma (58%) albergan al menos una variante rara en cualquiera de los genes incluidos en el panel. Los genes con mayor número de variantes alélicas raras fueron ATM y NF1 (15% y 14% respectivamente), seguidos de TIAM1 y ALK (11% cada uno). Interesantemente, el estudio de supervivencia de los pacientes reveló que las variantes genéticas detectadas en el gen TIAM1 se asociaban con un pronóstico de la enfermedad más favorable, tanto en supervivencia global como en tiempo libre de progresión/recaída, resultando además ser una variable de pronóstico independiente.
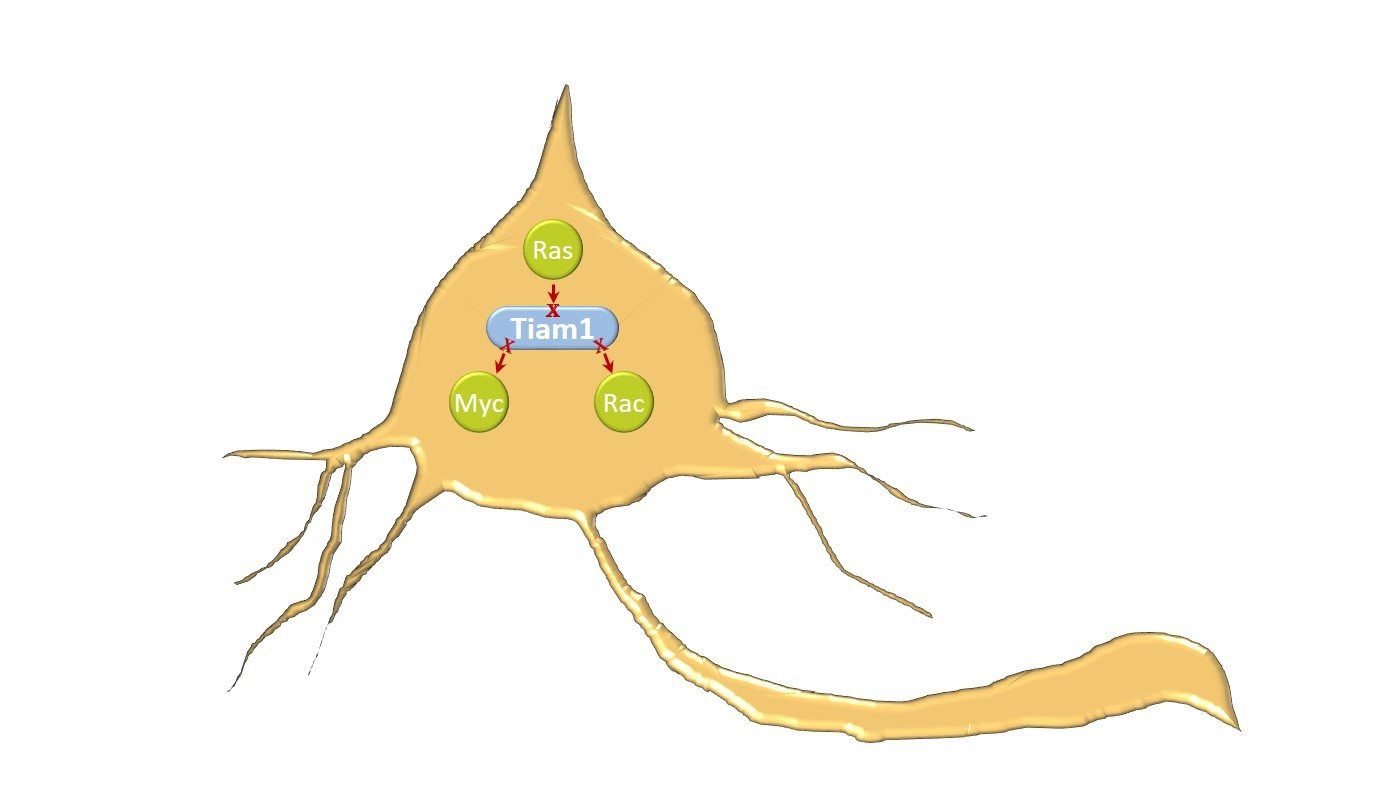
Para comprender el papel de las variantes de TIAM1 en la etiología y progresión del neuroblastoma, se evaluó su efecto en los dominios de señalización de la proteína TIAM1 así como las características clínicas de cada paciente. Las variantes identificadas se distribuyen principalmente en tres regiones de señalización: 1) la región N-terminal que permite la unión de miembros de la familia MYC para promover la co-activación transcripcional e inhibir la apoptosis celular, 2) el dominio de unión a RAS (RBD) para transducir su señalización a efectores aguas abajo, y 3) el dominio catalítico (DH-PH2) que permite la activación de RAC y de este modo, activa la migración celular y la neuritogénesis. Así pues, nuestros hallazgos sugieren que estas variantes alélicas inducen la pérdida de función parcial o completa de la proteína TIAM1 en las redes de señalización oncogénica relacionadas con las GTPasas RAS y RAC así como con MYC, resultando ser de esta forma, variantes protectoras que se asocian con un mejor pronóstico de la enfermedad.
Las variantes de TIAM1 se encontraron tanto en pacientes clasificados en el grupo de riesgo alto como en el de riesgo intermedio/bajo. Además, la coexistencia de variantes de TIAM1 con otras alteraciones genéticas asociadas con mayor riesgo resultó ser frecuente: 3 casos presentaban también amplificación de MYCN, 2 casos tenían variantes concomitantes en ALK y 1 en NF1. Aun así, la presencia de variantes en TIAM1 resulta de mejor pronóstico independientemente de la concomitancia con otras alteraciones genéticas de conocido carácter patogénico. Curiosamente, ninguno de los pacientes en los que se identificaron variantes de TIAM1 había fallecido, y solamente uno de ellos había presentado recaída/progresión de la enfermedad. Además, la secuenciación del tumor en paralelo con la sangre periférica reveló la presencia de estas variantes en la línea germinal en al menos 5 de los 12 casos, lo que apunta que muchas de estas variantes raras en TIAM1 pudieran ser germinales.
Todos estos resultados sugieren que el señalosoma controlado por TIAM1 puede ser esencial para el desarrollo del neuroblasoma y que su inhibición podría ser una nueva diana terapéutica que ayude a mejorar la eficacia del tratamiento convencional en este tipo de cáncer. El siguiente paso sería incorporar estos estudios a la práctica clínica para mejorar las herramientas y los procedimientos de diagnóstico.
Referencia: Sanmartín E, et al. TIAM1 variants improve clinical outcome in neuroblastoma. Oncotarget. 2017 Apr 3. doi: http://dx.doi.org/10.18632/oncotarget.16787

