
Silvia Martín-Almedina1 e Inés Martínez-Corral2
1 Lymphovascular Research Unit, Cardiovascular and Cell Sciences Institute, St. George’s University of London, London, United Kingdom. 2 Department of Immunology, Genetics and Pathology, Uppsala University, Uppsala, Sweden.
La hidropesía fetal (hydrops fetalis) se caracteriza por una acumulación excesiva de líquido intersticial generalizada, o presente en dos o más cavidades corporales del feto o del recién nacido incluyendo abdomen, pleura y pericardio. Se puede clasificar en dos tipos dependiendo de su origen: hidropesía fetal inmunitaria y no inmunitaria. La primera se desarrolla frecuentemente como consecuencia de una incompatibilidad de los sistemas Rh materno-fetales, originando una reacción adversa del sistema inmune. Sin embargo, la mayoría de los casos (85%) es de origen no inmunitario y de éstos, el 20% es de origen desconocido. Aproximadamente el 15% de los casos de hidropesía fetal no inmunitaria van asociados a una disfunción del sistema linfático (LRHF, del inglés Lymphatic-related (non-immune) hydrops fetalis). Esta manifestación de la patología ha sido categorizada a su vez en un subgrupo de linfedemas primarios conocido como displasia linfática generalizada (GLD, del inglés Generalized Lymphatic Dysplasia) y que se define como un linfedema con afectación sistémica o visceral que incluiría la LRHF (incluso si el linfedema no está completamente extendido). LRHF/GLD va frecuentemente asociada a abortos espontáneos o la muerte del recién nacido por lo que la identificación de mutaciones genéticas causantes de esta patología es el primer paso para lograr mejores pruebas diagnósticas, asesoramiento genético y tratamientos más refinados para tratar la enfermedad en el futuro.
En nuestro trabajo hemos estudiado dos familias con un historial de LRHF/GLD con una elevada incidencia de muertes in-utero o neonatales. La evaluación clínica en éstas dos familias dictaminó hidropesía fetal prenatal o hidrotórax neonatal (acumulación de líquido en el espacio pleural) de severidad variable así como una elevada incidencia de abortos espontáneos durante el primer trimestre. Generalmente, esta patología se asocia con una alta mortalidad, sin embargo, los recién nacidos que sobrevivieron el periodo neonatal mostraron una mejoría en el tiempo, con una resolución espontánea del edema y de las efusiones pleurales. Solo 1 de 5 adultos desarrolló linfedema en las extremidades inferiores en la adolescencia y 3 de 5 adultos presentaban varices. Muestras procedentes de ambas familias fueron sometidas a técnicas masivas de secuenciación de DNA con el fin de identificar posibles mutaciones relacionadas con la enfermedad. Los resultados obtenidos mostraron que genes relacionados hasta el momento con la presencia de linfedema (CCBE1, VEGFR3 y VEGFC) no presentaban ningún tipo de variación; sin embargo, se identificaron dos mutaciones puntuales (ambas en heterocigosis) en el gen EPHB4 (c.2216G>A, p.Arg739Glu y c.2345T>G, p.Ile782Ser) que no habían sido descritas anteriormente en las bases de datos públicas y que no aparecen en un total de 900 controles incluidos en el ensayo. Concluimos por tanto, que estas mutaciones son posiblemente las causantes del fenotipo observado en pacientes con LRHF/GLD independientemente de la severidad de éste.
EPHB4 codifica para un receptor con actividad tirosina-quinasa y la unión con su ligando Ephrin B2 induce la activación de diferentes cascadas de señalización celular. Las dos mutaciones identificadas en EPHB4 se encuentran localizadas en residuos altamente conservados de su dominio tirosina-quinasa y afectarían por lo tanto a la capacidad del receptor de activarse tras la unión a su ligando. La sobre-expresión de las proteínas portadoras de las mutaciones en células endoteliales linfáticas confirmó que la actividad tirosina-quinasa de EPHB4 tras la activación con Ephrin B2 se ve anulada, indicando que la disminución de la actividad del receptor contribuiría al desarrollo de la patología.
La vía de señalización Ephrin B2/EPHB4 es crítica para el desarrollo del sistema cardiovascular durante la embriogénesis. Tanto Ephrin B2 como EPHB4 son también esenciales para la remodelación de los vasos linfáticos y la formación de las válvulas linfáticas durante el desarrollo postnatal temprano. Sin embargo, no hay nada descrito sobre su papel durante la formación del sistema linfático que pudiese explicar el efecto de estas mutaciones en pacientes con LRHF/GLD. El siguiente paso del estudio consistió por tanto en analizar mediante la utilización de modelos genéticos de ratón, el efecto de la inactivación de Ephb4 específicamente en células endoteliales linfáticas durante una etapa temprana del desarrollo embrionario. A tal fin utilizamos la combinación de tres modelos (Prox1-creERT2, EphB4fl y R26-mTmG) que nos permitieron inactivar Ephb4 exclusivamente en células endoteliales linfáticas de una forma inducible (mediante administración de tamoxifeno) en momentos puntuales del desarrollo embrionario, y a su vez, marcar estas células mediante la proteína verde fluorescente (GFP). La inactivación de Ephb4 durante los días de desarrollo embrionario 10 (E10)- E14 dio lugar a una alta proporción de embriones (Ephb4fl/fl,Prox1-CreERT2,R26-mTmG) con edema subcutáneo y sangre en el interior de los vasos linfáticos a E15.5. El análisis de la piel de estos embriones puso de manifiesto además vasos linfáticos dilatados y tortuosos en comparación con los vasos linfáticos de embriones control. Sin embargo, cabe destacar que estos vasos linfáticos anormales no presentaban una alta proporción de células GFP+ (células dónde Ephb4 ha sido inactivado), indicando que la dilatación y el contenido en glóbulos rojos podría ser un efecto secundario. Apoyando esta teoría, la inactivación de Ephb4 desde E12.5 no mostró ningún tipo de fenotipo en los embriones analizados a E15.5, lo que sugirió un papel fundamental de Ephb4 en el sistema linfático durante los días E10-E12 de desarrollo embrionario.
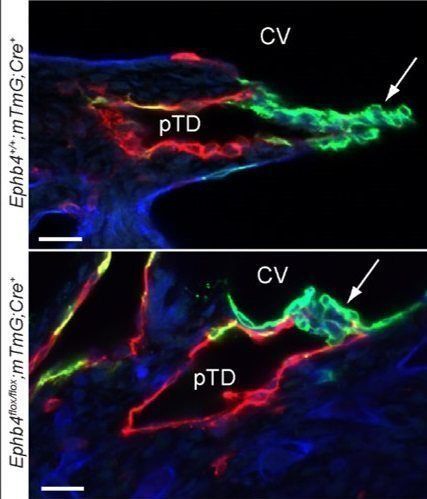
Durante los días E10-E13 de desarrollo embrionario es cuando se produce la formación de las válvulas linfático-venosas (LVVs, lymphovenous valves) que separan el sistema linfático del sanguíneo. Con el fin de analizar el papel de Ephb4 en la formación de estas válvulas indujimos la inactivación del receptor a E10-E12, y analizamos la estructura de las LVVs en los animales a E13. Nuestros resultados muestran que la inactivación del receptor Ephb4 durante este momento del desarrollo embrionario da lugar a la malformación de las LVVs, que en lugar de mostrar dos valvas orientadas hacia el interior de la vena cardinal, muestran una acumulación anormal de células endoteliales linfáticas GFP+ (células con Ephb4 inactivo). El mal funcionamiento de estas válvulas explicaría el fenotipo observado en los vasos linfáticos de los embriones analizados.
En resumen, en nuestro trabajo hemos identificado 2 mutaciones inactivadoras de la actividad tirosina-quinasa en el gen EPHB4, en 11 individuos procedentes de 2 familias con LRHF/GLD. Es la primera vez que se describe una forma autosómica dominante de esta patología, que presenta una manifestación variable del fenotipo. Hemos utilizado un modelo genético murino para demostrar que la deficiencia de Ephb4 específicamente en el endotelio linfático durante el desarrollo embrionario temprano da lugar a la malformación de las válvulas que separan el sistema linfático del sanguíneo, y esto puede contribuir a la aparición de edema en pacientes con LRHF/GLD. Nuestro trabajo relaciona, por primera vez, una manifestación clínica en humanos con mutaciones en EPHB4.
Referencia: Martin-Almedina S and Martinez-Corral I, et al. EPHB4 kinase-inactivating mutations cause autosomal dominant lymphatic-related hydrops fetalis. J Clin Invest. 2016 Jul 11. pii: 85794. doi: 10.1172/JCI85794.

