Autores: David Pla-Martín#, Eduardo Calpena#, Vincenzo Lupo, Celedonio Márquez, Eloy Rivas, Rafael Sivera, Teresa Sevilla, Francesc Palau§, Carmen Espinós§
#,§Estos autores han contribuido por igual al manuscrito
La neuropatía de Charcot-Marie-Tooth (CMT) es una enfermedad rara y sin embargo, se trata de una de las enfermedades neurológicas hereditarias más frecuentes. Bajo el nombre de CMT se engloba a un conjunto clínico y genéticamente heterogéneo de neuropatías, con varios tipos y subtipos, que se caracterizan por afectar al sistema nervioso periférico. Clínicamente la enfermedad de CMT cursa con debilidad progresiva y atrofia en los músculos distales principalmente de las extremidades inferiores, que se presenta con déficit motor y en menor grado, déficit sensitivo. Debido a la gran heterogeneidad clínico-genética se pueden establecer clasificaciones de diferentes formas de CMT atendiendo a criterios electrofisiológicos e histopatológicos (formas desmielinizantes o CMT1, formas axonales o CMT2, y formas intermedias o I-CMT) o considerando el patrón de herencia y en función de los genes causantes de la enfermedad, ya que hasta la fecha se han descrito más de 50 genes/loci implicados en CMT.
Algunas formas CMT se caracterizan por presentar penetrancia incompleta y expresividad variable. Este es el caso de la forma autosómica dominante CMT2K causada por mutaciones en el gen GDAP1 que cursa con una gran variabilidad intrafamiliar: es posible encontrar desde portadores asintomáticos hasta enfermos con un cuadro clínico grave en una misma familia siendo todos ellos portadores de la misma mutación en GDAP1. Es en este contexto donde se pone de manifiesto la existencia de modificadores genéticos que pueden contribuir a las consecuencias fenotípicas de las mutaciones causales y justificar en mayor o menor grado, la variabilidad clínica observada.
En un estudio colaborativo entre varios grupos españoles, se ha identificado el gen juntofilina-1 (JPH1) como modificador de la expresión clínica de CMT causada por mutaciones en el gen GDAP1.
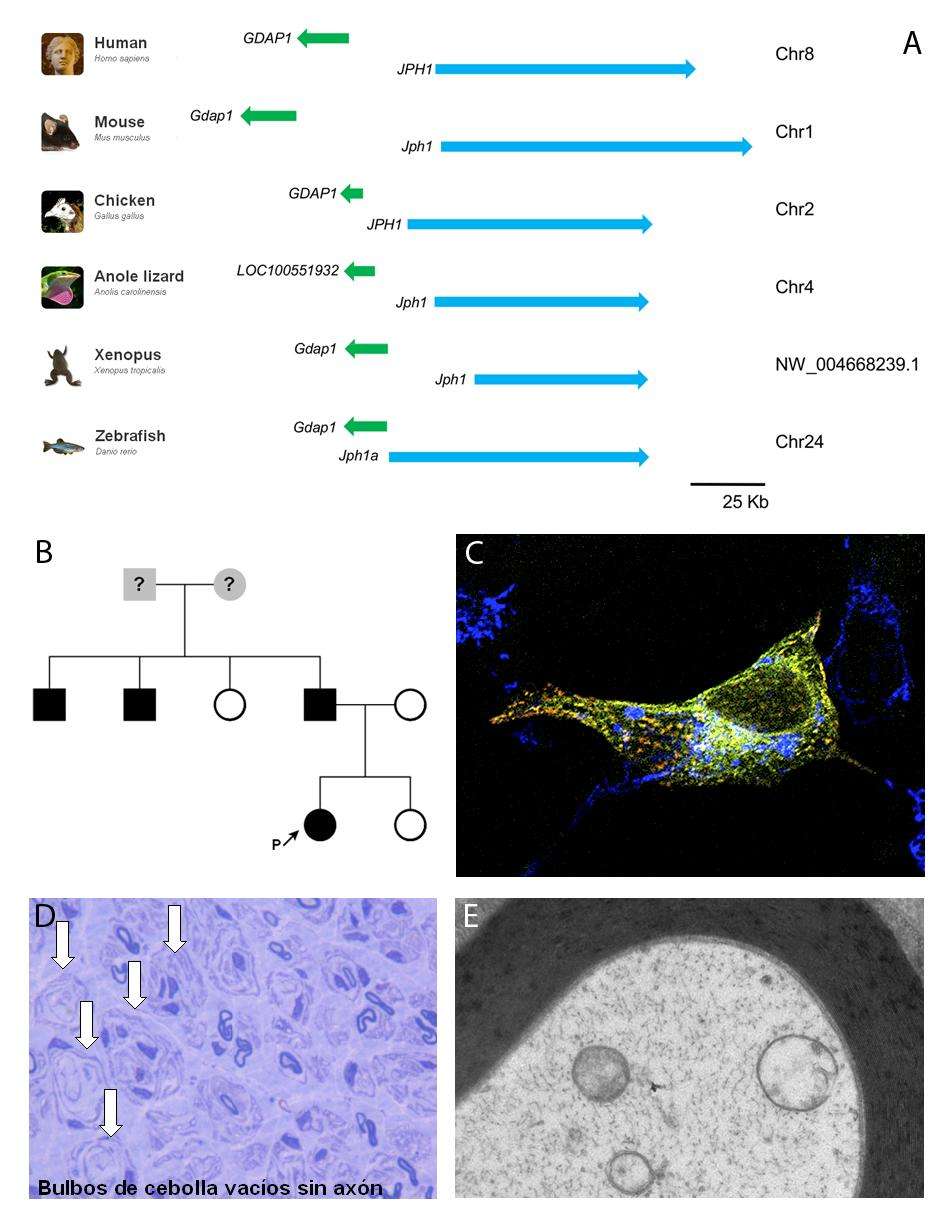
El déficit de GDAP1 en células provoca un defecto en un mecanismo conocido como SOCE (store-operated calcium entry), que es fundamental para una correcta homeóstasis del calcio celular. Tomando este hecho como punto de partida para la búsqueda de posibles modificadores que pudieran modular este fenotipo, los investigadores centraron su atención en el gen JPH1 por ser un candidato idóneo: (a) por un lado, se trata de un gen que en vertebrados mantiene una estrecha relación genómica con el gen GDAP1, ya que en un estudio de genómica comparativa se demuestra que estos genes en vertebrados se han conservado juntos, vecinos en el genoma y, (b) por otro lado, al igual que ocurre con GDAP1, JPH1 está implicado en el SOCE. Estos datos sugerían que ambos constituyen un cluster conservado de genes funcionalmente relacionados.
Usando como modelo las células deficientes de GDAP1, los investigadores han sido capaces de rescatar el defecto en el SOCE que presentaban estas células mediante la sobreexpresión de JPH1. Habiendo demostrado que JPH1 es un modificador funcional de GDAP1, la siguiente cuestión a contestar era si es posible que mutaciones/polimorfismos en JPH1 pudieran explicar casos de variabilidad intrafamiliar de CMT2K.
Para responder a esta cuestión se realizó un cribado mutacional del gen JPH1 en una serie de pacientes con CMT2K portadores de la mutación GDAP1 p.R120W que es la más prevalente asociada a esta forma clínica. En este estudio identificaron un paciente portador del cambio JPH1 p.R213P que presenta un cuadro clínico más grave en comparación con otro miembros afectados en su familia, cuando todos ellos son portadores de la mutación GDAP1 p.R120W.
Para tratar de demostrar que esta variante de JPH1 pudiera contribuir a la expresividad clínica del paciente, se llevaron a cabo estudios celulares. De entre los hallazgos obtenidos destaca que la variante JPH1 p.R213P es incapaz de recuperar el defecto en el SOCE de las células deficientes de GDAP1; pero además, cuando en células control se combinan las mutaciones GDAP1 p.R120W y JPH1 p.R213P, se produce una reducción drástica del SOCE. Estos resultados apoyan al posible efecto de la mutación de JPH1 como modulador negativo y que tendría consecuencias en presencia de defectos en GDAP1.
Los datos obtenidos en este trabajo, en su conjunto, han permitido establecer una relación funcional entre JPH1 y GDAP1, y más en concreto, que JPH1 actuaría como modificador negativo de GDAP1, agravando la clínica que estos pacientes manifiestan.
Referencia:
Pla-Martín D, Calpena E, et al. Junctophilin-1 is a modifier gene of GDAP1-related Charcot-Marie-Tooth disease. Hum Mol Genet. 2014 Aug 28. pii: ddu440.
Afiliaciones:
Programa científico de Enfermedades Raras y Genéticas. Centro de Investigación Príncipe Felipe (CIPF). Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER): David Pla-Martín, Eduardo Calpena, Vincenzo Lupo, Francesc Palau, Carmen Espinós
Departamento de Neurología y departamento de Patología. Hospital Universitario Virgen del Rocío: Celedonio Márquez, Eloy Rivas
Departamento de Neurología. Hospital Universitari i Politècnic La Fe. Instituto de investigación Sanitario (IIS)- La Fé. Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED): Rafael Sivera, Teresa Sevilla
Facultad de Medicina. Universidad de Castilla la Mancha: Francesc Palau
Departamento de Genética. Universitat de València : Carmen Espinós