
Amparo Tolosa, Genética Médica News
Todas las células somáticas de un organismo contienen el mismo genoma. Sin embargo, cada tipo celular puede presentar un perfil de expresión génica diferente, en función de cómo interprete las instrucciones genéticas marcadas por la secuencia de su ADN. Las claves para interpretar o ejecutar estas instrucciones son determinadas por el epigenoma, el conjunto de elementos funcionales – marcas bioquímicas o fragmentos de ARN – que regulan la expresión génica sin modificar la secuencia de ADN.
Los mapas epigenómicos de referencia de más de 100 tipos celulares y tejidos fueron recientemente publicados a través del Programa del Mapa Epigenómico. Continuando con la estrategia de analizar el epigenoma del cuerpo humano y determinar su influencia en la diversidad entre los diferentes órganos y tejidos, un trabajo del Instituto Salk de Estudios Biológicos, en EE.UU., ha obtenido los mapas de metilación, una de las marcas epigenéticas más frecuentes, de diferentes órganos obtenidos de donantes individuales y combinado la información epigenética con la referente a la secuencia genómica y expresión génica global.
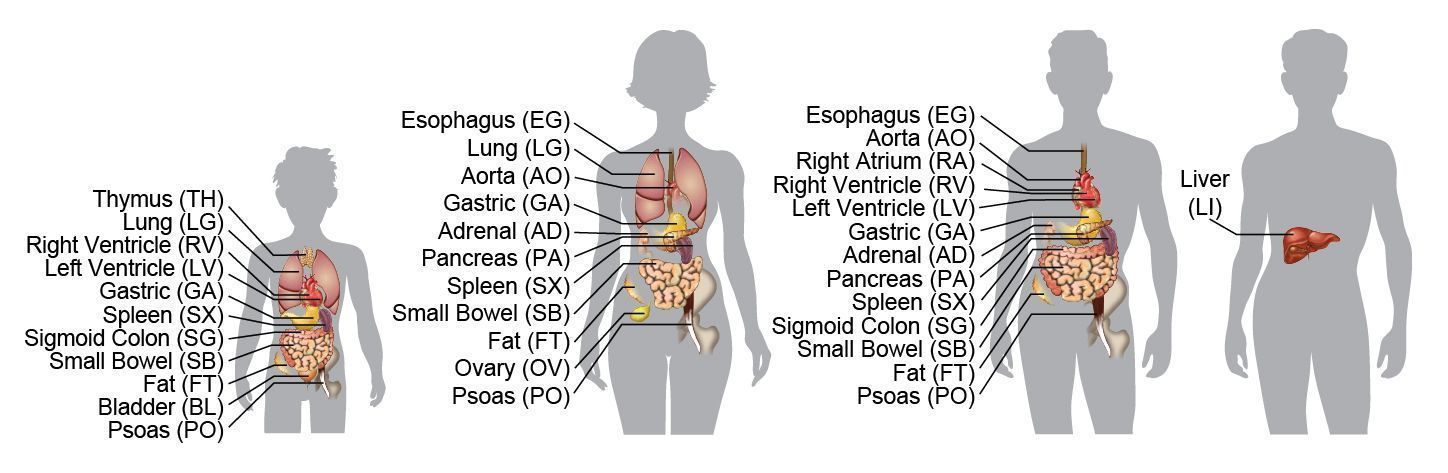
“Hemos encontrado que no todos los órganos que investigamos son iguales en términos de patrones de metilación,” indica Joseph R. Ecker, director del Laboratorio de Análisis Genómico del Instituto Salk y uno de los autores principales del trabajo. “Las firmas de metilación son lo suficientemente diferentes entre órganos como para observar los patrones de metilación de un tejido y saber si se trata de músculo, timo o páncreas.”
En las regiones que muestran diferencias en la metilación entre los órganos analizados, que incluyen tanto los denominados “promotores” de los genes como otras regiones, los investigadores observaron una correlación entre los niveles de metilación y la expresión de los genes cercanos. Concretamente, a mayor metilación se observó menor expresión de los genes, lo que apoya la consideración general de que las marcas de metilación suelen actuar como señales indicadoras para que la maquinaria de expresión génica no se active en los genes donde se encuentran.
En diversos tejidos analizados, los investigadores detectaron una cantidad apreciable de metilación fuera de las típicas regiones ricas en dinucleótidos CG, algo no esperado y que sólo había sido observado en el contexto de ciertos tipos celulares como las células madre embrionarias humanas o las células del cerebro. El análisis de este tipo de metilación reveló una distribución específica de tejido y mostró que los genes afectados están involucrados en diferentes funciones. Además, con excepción de las células cerebrales, la metilación en los sitios ajenos a las regiones CG disminuye durante la diferenciación en los diferentes tipos celulares especializados, apuntando a que este tipo de metilación es represiva. El equipo de investigadores opina que estos resultados abren la posibilidad de que la metilación en sitios diferentes a CG actúe en las células madre adultas para reprimir ciertos genes durante la transición al estado diferenciado.
Por último, utilizando los patrones de metilación los investigadores llevaron a cabo predicciones de genes que se escapan a la inactivación del cromosoma X en tejidos específicos.
En el trabajo del equipo de Ecker se analizó la metilación, expresión y secuencia del genoma en muestras postmortem de diferentes personas sanas, dos hombres, una mujer y un niño. La idea de obtener perfiles epigenómicos de personas sanas, está apoyada por la consideración de que diferentes enfermedades o patologías pueden ser causadas o mostrar una alteración en los patrones epigenéticos de las células afectadas. Disponer de los mapas epigenéticos y entender su participación para el correcto funcionamiento de cada tipo celular es como disponer de las claves para interpretar el libro de instrucciones del organismo. Y lo que es igual de importante, contribuye a facilitar qué sucede en las situaciones patológicas y como esta información podrá ser utilizada en el futuro con fines diagnósticos o pronósticos. “Se puede imaginar que eventualmente, si alguien tiene un problema, una biopsia no buscará únicamente caracterizar las células o los genes, sino también el epigenoma,” manifiesta Ecker.
Referencia: Schultz MD, et al. Human body epigenome maps reveal noncanonical DNA methylation variation. Nature. 2015 Jun 1. doi: 10.1038/nature14465.
Fuente: http://www.salk.edu/news/pressrelease_details.php?press_id=2085

