
Eduardo Andrés-León 1,† , Ildefonso Cases2, Sergio Alonso3 y Ana M. Rojas1
1Grupo de Biología Computacional y Bioinformática. Instituto de Biomedicina de Sevilla.
2REDgene Bioinformatics, Sevilla.
3Programa de medicina personalizada y preventiva del cáncer. (PMPPC), Germans Trias I Pujol (IGTP), Campus Can Ruti, Badalona.
†Dirección actual: Unidad de Bioinformática, Instituto de Parasitología y Biomedicina “López-Neyra”, Consejo Superior de Investigaciones Científicas (IPBLN-CSIC), Granada.
El reciente desarrollo de potentes tecnologías de secuenciación ha facilitado abordar varias preguntas de gran interés para la investigación oncológica. En primer lugar, determinar la contribución de los microARNs en la regulación de la expresión de genes relevantes en el desarrollo tumoral y, en segundo lugar, si estas asociaciones microARN-genes tienen un valor pronóstico para la supervivencia de los pacientes.
Numerosos estudios previos han demostrado que los microARNs son moduladores de la expresión génica y que su desregulación juega un importante papel en el desarrollo tumoral. Sin embargo, estos estudios han sido siempre abordados de manera independiente en determinados tipos tumorales. Existen también estudios describiendo determinadas firmas de expresión de microARNs con valor pronóstico pero, de nuevo, limitados a ciertos tipos de tumores.
Hasta la fecha, ningún estudio ha abordado el análisis de pares constituidos por microARNs/genes diana como potenciales indicadores pronósticos en una larga batería de tipos tumorales distintos. Un estudio de estas características presenta una complejidad excepcional debida a la reducida especificidad de estas moléculas de ARN que en muchos casos regulan numerosos genes, que estos genes pueden ser regulados por otros mecanismos (metilación, ganancias y pérdidas cromosómicas) y que a su vez pueden ejercer efectos pleiotrópicos en redes de regulación, y sumado a todo ello la complejidad intrínseca en el modelado de estas redes, de las que hay muy poca información.
En este marco nuestro estudio postula una hipótesis principal relativamente simple, teniendo en cuenta siempre las limitaciones mencionadas anteriormente: que el silenciamiento de ciertos genes diana está mediado principalmente por la sobreexpresión de microARNs, en lo que hemos denominado pares miARN-ARNm, y que este silenciamiento tiene un efecto fenotípico predecible, en particular una asociación con la supervivencia de los pacientes.
Para este estudio, la disponibilidad de datos de secuenciación masiva ha sido fundamental con el objeto de identificar tendencias globales en un conjunto muy grande de datos, donde el análisis integrativo ha permitido el abordaje de nuestra hipótesis a gran escala. Para ello, hemos utilizado datos procedentes del atlas genómico del cáncer (TCGA, del inglés The Cancer Genome Atlas), incluyendo datos de acceso controlado, y que contienen información referente a numerosas características asociadas a miles de muestras pertenecientes a distintos tipos tumorales.
En nuestro estudio, hemos seleccionado 15 tipos de cáncer que contenían información simultánea sobre expresión de genes y de microARNs, datos de metilación y alteraciones en el número de copias (ganancias y pérdidas cromosómicas). Nuestro estudio analiza más de 18.000 muestras procedentes de esos 15 tipos tumorales diferentes.
Como primer filtro, seleccionamos siete rutas de señalización relevantes para la carcinogénesis, reduciendo así el ruido transcripcional y enfocándonos directamente en un contexto relacionado con mecanismos moleculares que dictaminan el desarrollo tumoral. La comparación de los niveles transcripcionales de los tumores con los tejidos no tumorales permitió identificar genes y microARNs diferencialmente expresados de una forma recurrente, es decir, desregulados en la mayoría de los tipos tumorales estudiados, descartando así alteraciones espúreas o aquellas que son exclusivas de un tipo particular de cáncer. Para identificar posibles pares miARN-ARNm entre los miARNs y ARNms desregulados, aplicamos un primer filtro basado en la predicción de los genes diana para cada miARN, generando así una lista de pares en los que la expresión del gen (ARNm) está, con cierta probabilidad, modulada por la expresión del miARN asociado.
Aplicamos posteriormente filtros adicionales para seleccionar un conjunto razonable de pares miARN-ARNm, simplificando la interpretación de los resultados. Dado que la hipótesis de partida era que el miARN regula negativamente la expresión de su gen/genes diana, seleccionamos aquellos pares que mostraban perfiles invertidos de desregulación, es decir, sobreexpresión del miARN acompañada de silenciamiento del ARNm o, inversamente, silenciamiento del miARN y sobreexpresión del ARNm. Así, obtuvimos una lista de 41 pares en los que la expresión del ARNm podría estar silenciada por el miARN asociado. Los resultados de nuestro análisis global replicaron estudios anteriores tanto a nivel del gen como a nivel del miARN.
Estas asociaciones se investigaron con mayor rigor mediante análisis de correlación lineal multifactorial que incluían ganancias o pérdidas cromosómicas, así como el estado de metilación, que pudieran afectar a la expresión de los genes diana.
De los 41 pares inicialmente identificados en los conjuntos diferencialmente expresados, 36 se retuvieron tras la inclusión de metilación, ganancias y pérdidas cromosómicas. 25 de ellos no han sido previamente identificados aunque, individualmente, cada miembro del par ya ha sido vinculado a determinados tipos tumorales de manera independiente. De estos 25 pares, 11 presentan una alta especificidad (es decir, pocos números de interactores para cada componente del par).
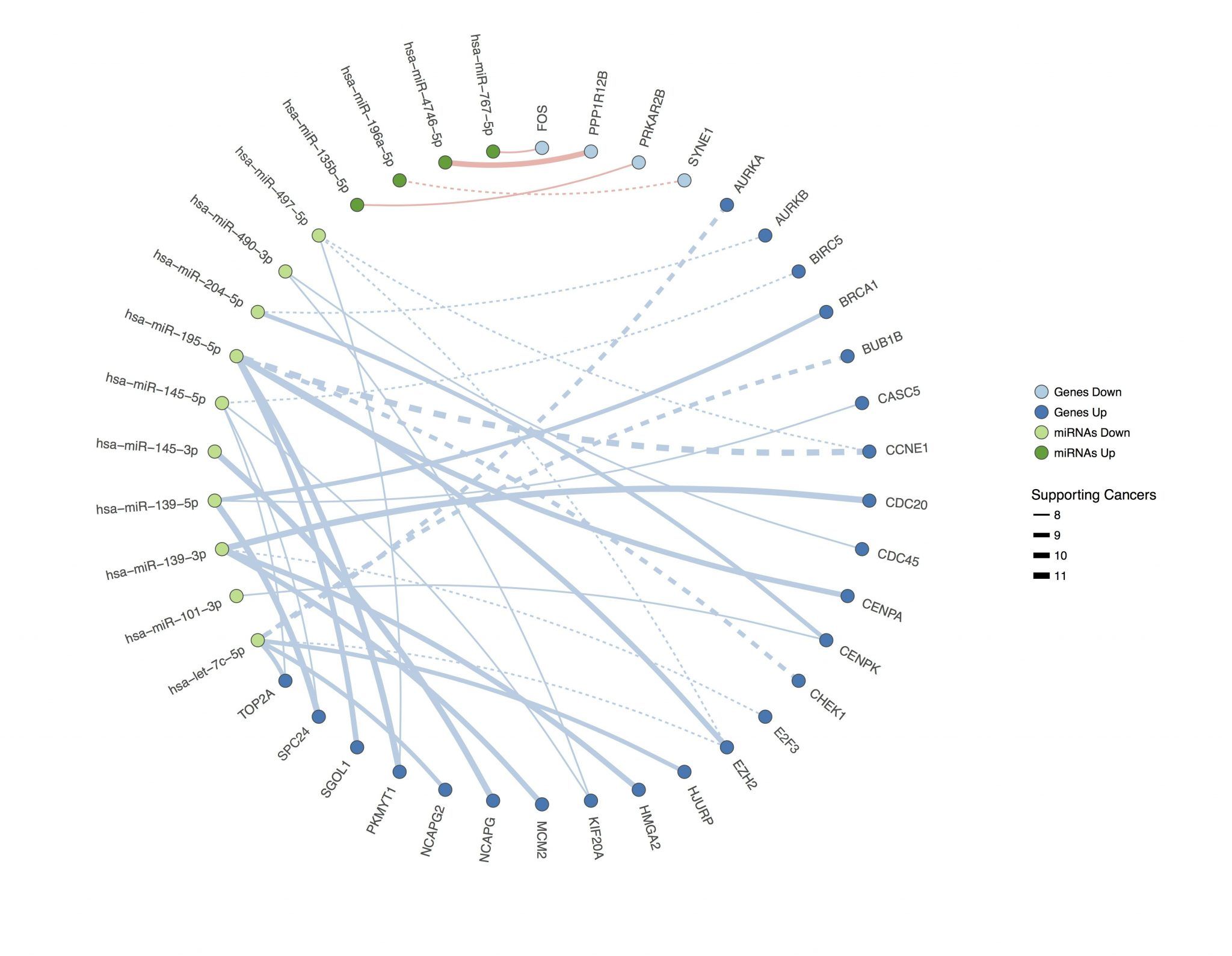
Identificamos, además, pares únicamente presentes en cánceres de pulmón de dos tipos histológicos diferentes, lo que indica un potencial valor como biomarcador en estos tipos tumorales.
Finalmente, determinamos el valor pronóstico de cada uno de estos pares en la supervivencia de los pacientes en un análisis global en el combinamos todos los tipos cáncer, y en análisis individuales en los que cada tipo de cáncer se estudio por separado, teniendo en cuenta siempre el tipo tumoral y el estadío de progresión, factores con un impacto fundamental en la supervivencia. Nuestros análisis revelaron la existencia de pares miARN-ARNm con valor pronóstico independiente del estadío en determinados tipos tumorales, que pueden mejorar los modelos predictivos y abrir nuevas vías de investigación para el futuro tratamiento del cáncer. Identificamos, además, genes pronósticos independientes de estadío no descritos anteriormente. Por ejemplo, CASC5 que ha sido descrito en cáncer de pulmón, nunca se había descrito asociado a cáncer de mama, tal y como nosotros hemos identificado en este estudio.
Resumiendo, este estudio puramente computacional describe un análisis exploratorio a gran escala que incluye la gestión de datos, conocimientos bioinformáticos, conocimiento de biología molecular y/o de biología del cáncer, seguidos de un análisis estadístico riguroso y posterior visualización de los datos, que pretende identificar dianas potenciales que tengan un posible valor terapéutico que puedan ser testadas experimentalmente.
Referencia: Andrés-León E, Cases I, Alonso S, Rojas AM. Novel miRNA-mRNA interactions conserved in essential cancer pathways. Sci Rep. 2017 Apr 7;7:46101. doi: http://dx.doi.org/10.1038/srep46101

