Xiomara Fernández Garibay, Juan Manuel Fernández Costa y Javier Ramón Azcón, Grupo de Biosensores para la Bioingeneieria. Instituto de Bioingeniería de Cataluña
Investigadores del Instituto de Bioingeniería de Cataluña en colaboración con el instituto de investigación sanitaria INCLIVA de Valencia han desarrollado el primer modelo 3D in vitro de músculo esquelético humano para la distrofia miotónica tipo 1 (DM1). El trabajo ha sido publicado en la revista Biofabrication, y demuestra el potencial de estos sistemas de cultivo 3D para el estudio preclínico de la distrofia miotónica.
La distrofia miotónica tipo 1 es una enfermedad genética altamente debilitante que afecta principalmente al músculo, causando degeneración y debilidad muscular. El síntoma definitorio de la enfermedad es la miotonía, que consiste en la incapacidad de relajar un músculo tras una contracción voluntaria. La DM1 es una condición multisistémica que también afecta otros tejidos como el cerebro y el corazón.
A pesar de ser una enfermedad rara, la DM1 es la miopatía hereditaria más frecuente en la población adulta. La causa molecular de esta enfermedad es una expansión del trinucleótido CTG en la región 3’ UTR del gen DMPK. En los transcritos de DMPK las repeticiones CUG adquieren forma de horquilla que secuestran proteínas de unión al ARN y se acumulan en el núcleo en forma de inclusiones ribonucleares o foci. Entre las proteínas secuestradas por los transcritos tóxicos DMPK se encuentran las proteínas de la familia Muscleblind-like (MBNL). La retención de MBNL en los foci provoca una falta parcial de su función, resultando en una alteración en los patrones de splicing que se relacionan directamente con varios síntomas característicos de la enfermedad.
En la actualidad no existe cura para la DM1. No obstante, el progreso en el descubrimiento de las causas moleculares de la enfermedad ha permitido el desarrollo de diferentes estrategias terapéuticas. Los modelos animales y los cultivos celulares tradicionales (en 2D) han sido esenciales para estos avances. Sin embargo, estos modelos carecen de relevancia fisiológica para los humanos. Por ejemplo, los cultivos celulares en 2D no representan la arquitectura compleja del músculo esquelético, compuesto por fascículos de fibras musculares alineadas, rodeadas de otras fibras y de matriz extracelular. Por otro lado, el uso de animales para experimentación farmacológica plantea varios dilemas éticos. Además, estos modelos in vivo han resultado no ser lo suficientemente efectivos en predecir la eficacia clínica de medicamentos, ya que es difícil extrapolar los resultados obtenidos con animales a seres humanos.
Para acelerar la investigación preclínica, los ensayos tradicionales podrían complementarse con modelos de tejidos 3D generados a partir de células de pacientes y que mimeticen la arquitectura del músculo esquelético (Fernández-Costa JM et al, 2021). Además, dada la gran variabilidad intrínseca de las distrofias musculares, el uso de células derivadas de pacientes ayudaría a que los resultados obtenidos tengan más relevancia para la práctica clínica.
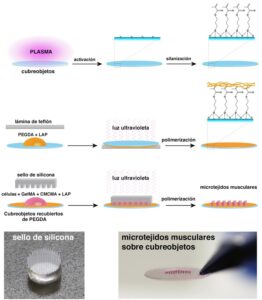
El tejido muscular esta formado por fibras alargadas y multinucleadas que se disponen de manera paralela. Durante el desarrollo, las células precursoras de músculo (mioblastos) se fusionan formando miotubos que después se convierten en miofibras maduras. Para obtener un modelo 3D de DM1 los investigadores mimetizaron esta estructura in vitro mediante un protocolo de micromoldeado (Figura 1). En este protocolo las células derivadas de pacientes fueron encapsuladas en micropatrones de un hidrogel fotopolimerizable. Los micropatrones confinan a los mioblastos topográficamente, promoviendo su alineación y fusión. Por otro lado, el hidrogel, una mezcla de gelatina metacrilada y celulosa metacrilada (GelMA-CMCMA), actúa como un andamio de matriz extracelular. El hidrogel de GelMA-CMCMA permite la adhesión celular mientras que retrasa la degradación enzimática de los micropatrones generando cultivos de larga duración, esenciales para plataformas de ensayos preclínicos.
Los investigadores obtuvieron microtejidos musculares 3D de DM1 (figura 2), los cuales retienen las características moleculares y estructurales de la enfermedad. Además, la encapsulación en micropatrones aumentó la formación de miotubos (evaluada por el índice de fusión) de DM1 en comparación a cultivos 2D, obteniendo valores similares a aquellos microtejidos sanos. Esto supone un cambio de paradigma para el estudio de fenotipos celulares para la DM1 ya que hasta el momento el índice de fusión se ha utilizado como fenotipo in vitro clásico de la enfermedad. Por otro lado, los autores analizaron el diámetro de los miotubos a partir de reconstrucciones 3D, observando que los miotubos enfermos tenían un diámetro significativamente menor a los miotubos sanos. En conclusión, los autores proponen que este fenotipo estructural mimetiza lo que ocurre en los músculos de los pacientes DM1, por lo tanto, es más relevante fisiológicamente y más interesante para evaluar posibles terapias para la DM1.
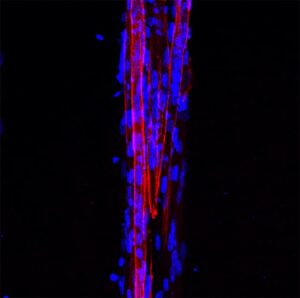
Para demostrar la aplicabilidad de este modelo in vitro en la evaluación de compuestos terapéuticos, los autores realizaron una prueba de concepto utilizando un oligonucleótido antisentido que previamente había desarrollado parte del equipo investigador. Concretamente usaron un oligonucleótido antisentido (antagomiRs) que bloquea específicamente el miRNA-23b y como consecuencia aumentan los niveles endógenos de las proteínas MBNL. El potencial terapéutico de antagomiR-23b se evaluó previamente en modelos celulares y animales para DM1 (Cerro-Herreros E et al, 2018 y 2020). En estos estudios se determinó que el tratamiento con el antagomiR-23b rescataba fenotipos moleculares y funcionales en estos modelos. En el presente trabajo, los microtejidos musculares de DM1 fueron tratados con antagomiR-23b. El tratamiento con el oligonucleótido aumentó los niveles de MBNL en los microtejidos. Además, tras el tratamiento, se observó un rescate de fenotipos moleculares y estructurales, incluyendo un incremento en el diámetro de los miotubos de DM1 hasta niveles de miotubos sanos.
Estos resultados demuestran que este nuevo modelo 3D creado a partir de células de paciente representa una valiosa alternativa al uso de animales o modelos 2D en la investigación preclínica. Además, este protocolo también podría aplicarse en el estudio de tratamientos para otras distrofias musculares.
Referencia: Fernández-Garibay X, et al. Bioengineered in vitro 3D model of myotonic dystrophy type 1 human skeletal muscle. Biofabrication. 2021, 13: 035035. doi: http://dx.doi.org/10.1088/1758-5090/abf6ae
Bibliografía:
Cerro-Herreros E, et al. miR-23b and miR-218 silencing increase Muscleblind-like expresión and alleviate myotonic dystrophy phenotypes in mammalian models. Nat. Commun. 2018, 9(1):2482. doi: http://dx.doi.org/10.1038/s41467-018-04892-4
Cerro-Herreros E, et al. Therapeutic Potential of AntagomiR-23b for Treating Myotonic Dystrophy. Mol. Ther. – Nucleic Acids. 2020, 837-849. doi: http://dx.doi.org/10.1016/j.omtn.2020.07.021
Fernández-Costa J.M., et al. Bioengineered in vitro skeletal muscles as new tools for muscular dystrophies preclinical studies. Journal of Tissue Engineering. 2021, 12. doi:http://dx.doi.org/10.1177/2041731420981339
Si te ha gustado esta noticia y quieres aprender más sobre Genética en Medicina, te interesan nuestros cursos y formación universitaria, así como nuestro canal audiovisual, Genotipia TV.