
Teresa Sevilla, Vincenzo Lupo, Dolores Martínez-Rubio, Paula Sancho, Rafael Sivera, María J. Chumillas, Mar García-Romero, Samuel I. Pascual-Pascual, Nuria Muelas, Joaquín Dopazo, Juan J. Vílchez, Francesc Palau, Carmen Espinós
La identificación de las bases genéticas que conducen a las enfermedades mendelianas raras hace posible el diagnóstico molecular y el consejo genético y también, contribuye a comprender los mecanismos de enfermedad, las rutas biológicas y la posibilidad de identificar posibles dianas terapéuticas. Hasta la fecha, se conocen las bases moleculares de menos de la mitad de las enfermedades raras genéticas. Las tecnologías Next Generation Sequencing (NGS) han creado nuevas e interesantes aproximaciones en los análisis genéticos trasladables al diagnóstico clínico, sobre todo la secuenciación de exomas.
La enfermedad de Charcot-Marie-Tooth (CMT) o neuropatía hereditaria sensitivo-motora (NHSM) es la enfermedad neurológica hereditaria más frecuente, aun siendo una enfermedad rara con una prevalencia de 28/100.000. Clínicamente cursa con una debilidad muscular distal progresiva y atrofia, pérdida sensorial distal con implicación de las extremidades superiores conforme la enfermedad progresa. Genéticamente se caracteriza por una amplia heterogeneidad con más de 50 genes implicados.
En algunos de los pacientes pertenecientes a la serie clínica de enfermos CMT que permanecían sin diagnóstico genético los investigadores secuenciaron el exoma. En una familia con CMT axonal se identificó una mutación novel c.568C>T (p.R190W) en el gen MORC2, un gen hasta la fecha no relacionado con neuropatías. Posteriormente, el análisis mutacional del gen en pacientes con CMT axonal pendientes de diagnóstico identificó dos casos esporádicos: uno de ellos con la misma mutación y otro con el cambio novel MORC2 p.S25L. Adicionales análisis genéticos e in silico han confirmado las mutaciones y predicho que son patológicas. MORC2 pertenece a la familia de proteínas microorchidia (MORC) y se ha postulado que participa en mecanismos de reparación de DNA. Hasta la fecha no se había relacionado MORC2 con neuropatías.

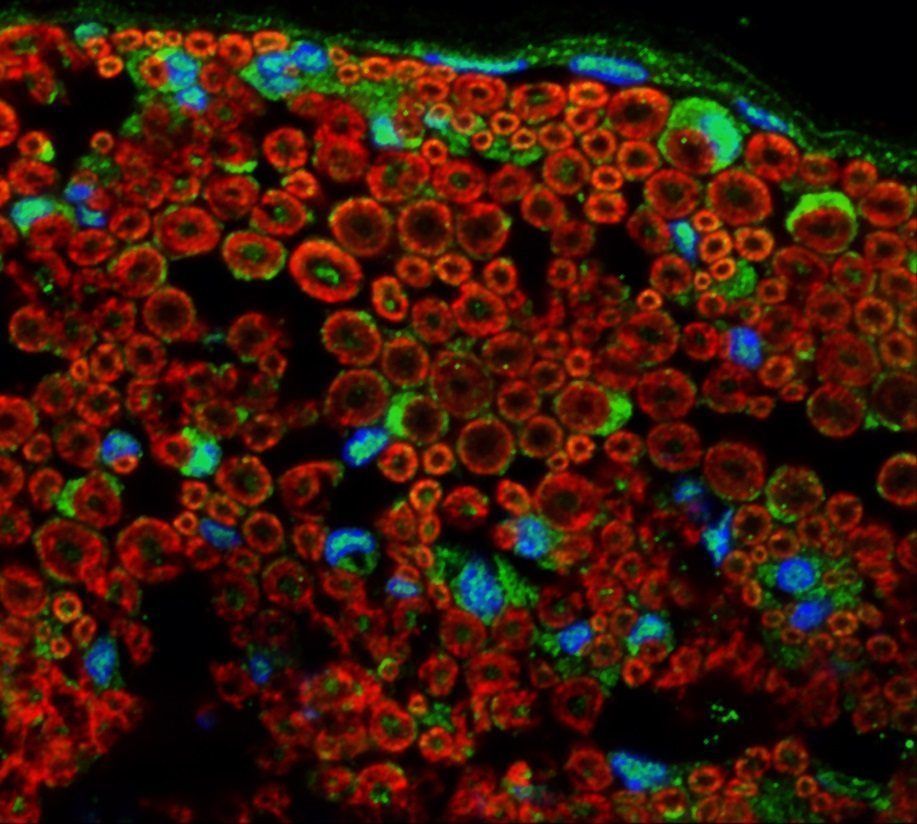
Los investigadores han demostrado que MORC2 se expresa en sistema nervioso periférico (PNS) de ratón (Figura 1). El gen se expresa en el axoplasma de los axones periféricos, así como también en células de Schwann. La expresión localizada en axón correlaciona perfectamente con la patología axonal que muestran los pacientes. El cuadro clínico es variable: debut congénito o infantil y un fenotipo similar a atrofia muscular espinal o inicio en la segunda década con calambres, debilidad distal y pérdida sensitiva. Las biopsias de nervio sural de los pacientes indican la existencia de una pérdida muy pronunciada de fibras axónicas (Figura 2) lo que concuerda con su fenotipo axonal.
En conclusión, MORC2 juega un papel en el sistema nervioso periférico, de modo que por primera vez en el grupo de neuropatías hereditarias sensitivo-motoras se describe un gen implicado en mecanismos de reparación de DNA que puede ser un nexo común a varias de las formas clínicas de este grupo de enfermedades, y por tanto, puede ser una diana terapéutica potencial que abre una ventana al posible tratamiento de este grupo de neuropatías hereditarias.
Referencia: Sevilla T, et al. Mutations in the MORC2 gene cause axonal Charcot-Marie-Tooth disease. Brain. 2015.doi: 10.1093/brain/awv311
Afiliaciones:
Programa científico de Enfermedades Raras y Genéticas. Centro de Investigación Príncipe Felipe (CIPF): Vincenzo Lupo, Dolores Martínez-Rubio, Paula Sancho, Carmen Espinós
Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER): Teresa Sevilla, Vincenzo Lupo, Dolores Martínez-Rubio, Paula Sancho, María J. Chumillas, Nuria Muelas, Juan J. Vílchez , Joaquín Dopazo, Francesc Palau, Carmen Espinós
Departamento de Neuropediatría. Hospital Universitario la Paz: Mar García-Romero, Samuel I. Pascual-Pascual
Departamento de Neurología. Hospital Universitari i Politècnic La Fe. Instituto de investigación Sanitario (IIS)- La Fé: Teresa Sevilla, Rafael Sivera, Nuria Muelas, Juan J. Vílchez
Departamento de Neurofisiología Clínica. Hospital Universitari i Politècnic La Fe. Instituto de investigación Sanitario (IIS)- La Fé: María J. Chumillas
Programa de Genómica Computacional. Centro de Investigación Príncipe Felipe (CIPF): Joaquín Dopazo
Departamento de Genética y Medicina Molecular. Institute for Rare Diseases (IPER), Hospital Sant Joan de Déu: Francesc Palau
Departamento de Medicina. Universitat de València: Teresa Sevilla, Juan J. Vílchez

