La epilepsia mioclónica progresiva constituyen un grupo de desórdenes familiares caracterizados por convulsiones mioclónicas (sacudidas repentinas e involuntarias de los músculos) ataxia, deterioro intelectual progresivo y degeneración neuronal. En su mayoría estos desórdenes se heredan de forma autosómica recesiva, con muy pocos casos de herencia autosómica dominante o mitocondrial y se han identificado diversos genes implicados en algunos de ellos. Sin embargo su diagnóstico clínico preciso es complicado, debido a la existencia de heterogeneidad genética y al solapamiento de los síntomas con otras patologías.
Un estudio colaborativo, en el que participan diferentes centros de investigación y hospitales de Europa, Norteamérica, Asia y Australia ha abordado el problema de la identificación de las causas moleculares de una serie de pacientes con epilepsia mioclónica progresiva recogidos durante los últimos 25 años.
Los investigadores secuenciaron el exoma, o parte del genoma que se traduce en proteínas, en 84 pacientes con epilepsia mioclónica progresiva sin causa identificada. Tras analizar las variantes genéticas y filtrarlas en función de su presencia en bases de datos o carácter patogénico, el equipo fue capaz de resolver cerca de un tercio de los casos.
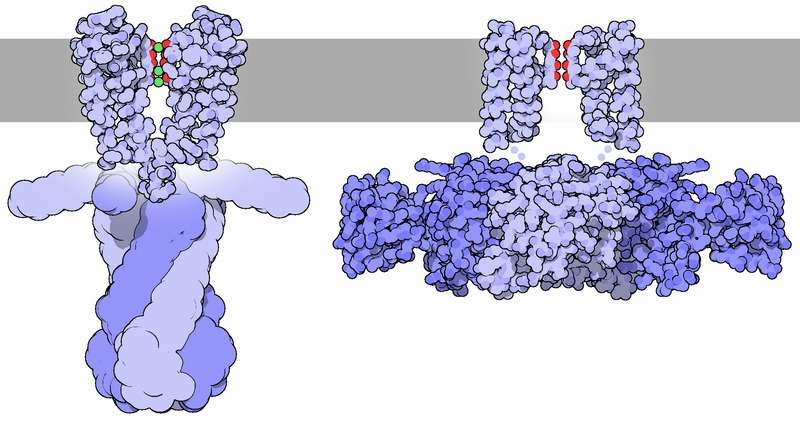
El resultado más sorprendente fue la presencia de una mutación de novo recurrente en el gen KCNC1 en 11 de los pacientes. La mutación, pArg329His, causa un cambio de aminoácido en una de las subunidades de la familia Kv3 de canales de potasio, en un dominio importante para su funcionamiento. Tanto la información obtenida por análisis bioinformáticos como los análisis funcionales en un modelo animal, Xenopus, indican que el cambio puede alterar la transmisión de señales de ciertas células nerviosas en las que se expresa el gen y con ello contribuir a las convulsiones. Otras evidencias apoyan también que la alteración de la función en las neuronas del cerebelo puede contribuir al daño motor.
Los investigadores indican que la recurrencia de la mutación, su elevada frecuencia de aparición de novo, no transmitida por sus padres, probablemente se debe a su localización, en una posición “caliente” para este tipo de cambios. La estimación de su recurrencia es que la mutación sucede en uno de aproximadamente 6 millones de concepciones.
Ante la participación de KCNC1 en la epilepsia mioclónica progresiva, se plantea la posibilidad de que la modulación farmacológica de su actividad podría ser una aproximación viable para el tratamiento de los pacientes con la mutación. No obstante, y a pesar de que existen fármacos antiepilépticos dirigidos hacia la regulación de otros canales iónicos similares, todavía no se dispone de ningún activador conocido para KV3.1.
Referencia:Muona M. A recurrent de novo mutation in KCNC1 causesprogressive myoclonus epilepsy. Nat Genet. 2014 Nov 17. doi: 10.1038/ng.3144.
Fuente: http://www.eurekalert.org/pub_releases/2014-11/uoh-ang111714.php