Mario Ezquerra*, Rubén Fernández-Santiago*
Laboratorio de Enfermedades Neurodegenerativas, IDIBAPS – Hospital Clínic de Barcelona, Universidad de Barcelona, CIBERNED
* ambos autores han contribuido en igual proporción al artículo
La enfermedad de Parkinson (EP) es una enfermedad neurodegenerativa frecuente con una prevalencia de 1/100 en personas mayores de 60 años. Alrededor del 10% de los casos son formas monogénicas causadas por mutaciones patogénicas que segregan con la enfermedad. Entre ellas, la EP causada por mutaciones en el gen LRRK2 es la más frecuente y más parecida clínico-patológicamente a la forma esporádica mostrando un patrón de herencia dominante pero con penetrancia incompleta. Por el contrario, el 90% de los casos son esporádicos y su causa es desconocida. El objetivo principal de nuestro trabajo fue abordar por primera vez si existían alteraciones epigenéticas asociadas con la EP, tanto en su forma monogénica asociada a LRRK2 como en la forma esporádica. Previamente, la epigenética había sido poco explorada en EP porque las neuronas dopaminérgicas (DAn), cuya neurodegeneración causa los síntomas motores clásicos, se localizan en un área del mesencéfalo ventromedial llamada substantia nigra, y esta región solamente es accesible post-mortem tras muchos años de enfermedad y alta tasa de pérdida neuronal (>70%).
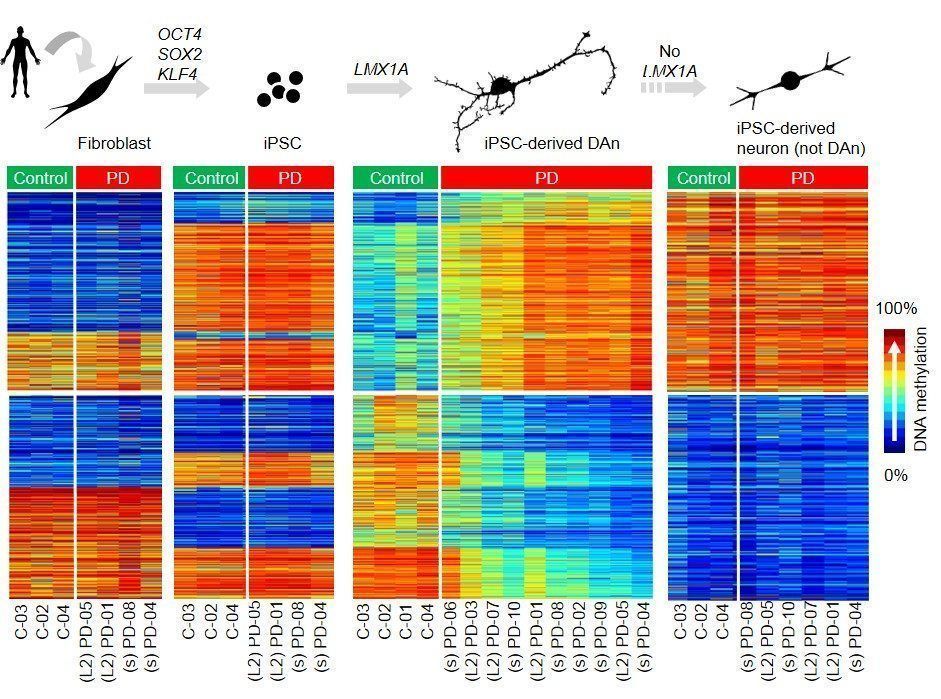
Para investigar la presencia de alteraciones epigenéticas en la EP, generamos DAn inducidas a partir de células somáticas de pacientes de EP incluyendo 4 pacientes con EP-LRRK2 causada por la mutación G2019S, 6 pacientes con EP esporádica y 4 controles sanos. Para generar DAn, usamos un protocolo previamente publicado (Sánchez-Danés et al., EMBO Mol Med 2012) que consistió en tres pasos: (i) biopsia de piel en el brazo de los sujetos y aislamiento de fibroblastos, (ii) reprogramación celular de fibroblastos a células madre pluripotentes inducidas (iPSC) mediante transducción retroviral de factores de reprogramación (OCT4, SOX2, KLF4) y (iii) diferenciación de iPSC a DAn mediante la transducción lentiviral del determinante dopaminérgico LMX1A. Usando este protocolo, observamos que las DAn resultantes eran morfológica y funcionalmente maduras, expresaban marcadores propios de DAn (TH, GIRK2 y FOXA2), y mostraban actividad eléctrica con potenciales de acción, de forma similar en casos y controles.
Mediante arrays de metilación de DNA de alta densidad (450k Illumina) estudiamos el metiloma de estas DAn inducidas. Al comparar el grupo de EP con controles identificamos por primera vez numerosas regiones diferencialmente metiladas que estaban asociadas con la enfermedad (2087 DMCpGs) (Fernández-Santiago et al., EMBO Mol Med 2015). Además, encontramos que la forma de EP monogénica LRRK2 y la forma esporádica eran similares y compartían patrones de metilación comunes (ver Figura 1). Por otra parte, al estudiar el metiloma de iPSC isogénicas de los mismos sujetos, o de fibroblastos parentales, no hallamos diferencias epigenéticas entre casos y controles. Estos resultados demostraron que las diferencias epigenéticas asociadas con la EP están latentes en células somáticas y se ponen de manifiesto solamente en el contexto celular dopaminérgico afectado por la enfermedad. Como experimento control, estudiamos el metiloma de cultivos neuronales isogénicos no enriquecidos en DAn generados mediante la no-transducción de LMX1A y tampoco observamos diferencias entre casos y controles. Este experimento demostró que las 2087 DMCpGs identificadas en EP eran atribuibles a las DAn de nuestros cultivos, pero no a otros tipos celulares o ni siquiera a otros tipos neuronales. Además, el patrón epigenético de las DAn de pacientes, pero no de controles, resultó ser más parecido al de cultivos neuronales no enriquecidos en DAn indicando una falta de identidad epigenética dopaminérgica propia de DAn sanas en la EP que podría estar relacionada con defectos durante el desarrollo neuronal en esta enfermedad.
En DAn observamos que la mayor parte de la hipermetilación diferencial de EP afectaba a enhancers (35%), que son regiones funcionalmente activas que pueden unir factores de transcripción (FT) para regular la expresión génica de sus genes diana. A su vez, usando datos de inmunoprecipitación de cromatina disponibles en el proyecto ENCODE, encontramos que los enhancers hipermetilados en EP mostraban enriquecimiento en lugares de unión de 23 FT. Entre éstos, hallamos una relación inversa significativa entre niveles de metilacion de enhancers y expresión de 4 FT clave que previamente habían sido relacionados con la EP (FOXA1, NR3C1, FOSL2 y HNF4A). Esta relación inversa consistía en déficits de expresión de los FT, tanto a nivel de RNA como de proteína, e hipemetilación de sus enhancer diana en las DAn de pacientes respecto a controles. Además, encontramos que la expresión de estos 4 FT estaba coordinada entre sí indicando que, globalmente, las diferencias epigenéticas observadas en EP estaban relacionadas con el déficit de una red de FT, más que de FT individuales.
Por último, también estudiamos el transcriptoma de las DAn e identificamos 437 genes diferencialmente expresados en EP respecto a controles, no hallando diferencias significativas de expresión entre EP-LRRK2 y EP esporádica. Los genes sobre-regulados en EP estaban relacionados con funciones neuronales, mientras que los infra-regulados con funciones homeostáticas básicas. Además, encontramos que los niveles de metilación de las 2.087 DMCpGs correlacionaban al menos en parte (17%) con los niveles de expresión de sus genes proximales. Es interesante mencionar que el conjunto de alteraciones epigenómicas, transcriptómicas, y los déficits de FT observados en nuestros cultivos de DAn de EP (30 días) anteceden en el tiempo la aparición espontánea de fenotipos asociados con la enfermedad en cultivos a largo plazo (75 días), tales como acumulación de a-sinucleina, déficits de autofagia, así como axo- y dendrogénesis reducidas en EP (Sánchez-Danés et al., 2012). Esta observación sugiere una posible relación causal entre cambios epigenéticos tempranos y fenotipos de EP tardíos.
En resumen, hemos identificado por primera vez alteraciones epigenéticas en el tipo celular principal afectado en esta enfermedad, hallando además que estas alteraciones son compartidas por los casos monogénicos LRRK2 y por los casos esporádicos. En el caso de los pacientes asociados a mutaciones en LRRK2, las alteraciones epigenéticas podrían explicarse por el impacto específico y patogénico de la mutación en las DAn. En los casos esporádicos, sin embargo, en los que se considera que la enfermedad es consecuencia de la interacción compleja del background genético y la exposición ambiental del individuo, es más difícil de explicar la presencia de estas alteraciones en las DAn, así que los fibroblastos originales podrían portar alteraciones moleculares resultantes de la genética e historia ambiental del paciente. Estas alteraciones moleculares no resultarían patogénicas en este tipo celular si no solamente en las DAn donde tendrían como consecuencia la aparición de defectos en el desarrollo epigenético dopaminérgico. Estos hallazgos señalarían a la EP como una enfermedad sistémica que afecta a otros tipos celulares no neuronales, aunque las DAn sean los tipos celulares vulnerables. Por otra parte, nuestro estudio pone en valor el uso de modelos neuronales derivados de iPSC no sólo para la investigación epigenética de la EP sino también potencialmente para modelar los efectos de factores ambientales en el epigenoma de pacientes. Nuestro estudio también sugiere que futuras estrategias terapéuticas basadas en el autotrasplante de células dopaminérgicas obtenidas a partir de iPSC en pacientes con EP habrían de asegurar previamente la correcta reprogramación epigenética de dichas células.
Referencia del artículo:
Fernández-Santiago R, et al. Aberrant epigenome in iPSC-derived dopaminergic neurons from Parkinson’s disease patients. EMBO Mol Med. 2015 Oct 29;7(12):1529-46. doi: 10.15252/emmm.201505439.
Abstract visual del artículo:
http://embopress.org/video_EMM-2015-05439
Literatura:
Sánchez-Danés A, et al. Disease-specific phenotypes in dopamine neurons from human iPS-based models of genetic and sporadic Parkinson’s disease. EMBO Mol Med. 2012 May;4(5):380-95. doi: 10.1002/emmm.201200215.
Schafer DP, Stevens B. Synapse elimination during development and disease: immune molecules take centre stage. Biochem Soc Trans. 2010 Apr;38(2):476-81. doi: 10.1042/BST0380476.
Petrucci S, Consoli F, Valente EM. Parkinson Disease Genetics: A «Continuum» From Mendelian to Multifactorial Inheritance. Curr Mol Med. 2014 Oct 10.

