- Nuevos modelos neurales de síndrome de Sanfilippo, útiles para estudiar potenciales tratamientos.
- El síndrome de Sanfilippo es una enfermedad rara caracterizada por una severa neurodegeneración para la que aún no existe tratamiento.
En los últimos años ha habido grandes avances en el campo de las enfermedades raras. Sin embargo, aquellas cuya principal sintomatología implica una alteración del sistema nervioso central siguen siendo un reto tanto en el desarrollo de potenciales terapias como de modelos de enfermedad que recapitulen adecuadamente esta degeneración.
Una de estas enfermedades raras con una clara patología neurológica es el síndrome de Sanfilippo (ver Benetó et al. 2020c y referencias incluidas en ese artículo), una mucopolisacaridosis (MPS) para la cual aún no existe un tratamiento efectivo. Está causada por mutaciones en distintos genes implicados en la ruta de degradación del heparán sulfato, cuyo acúmulo en los lisosomas causa la enfermedad. Según el gen mutado se reconocen 4 subtipos de la enfermedad (A-D), siendo los genes NAGLU y HGSNAT los alterados en los subtipos B y C, respectivamente.
Los modelos celulares y animales son de gran utilidad en el estudio de estas enfermedades. Sin embargo, cuando éstas implican alteraciones neurológicas la mayoría de los modelos existentes son humanos pero no neuronales, o neuronales pero no humanos, normalmente murinos.
En nuestro grupo se generaron previamente diferentes líneas de células madre pluripotentes inducidas (iPSCs), derivadas de fibroblastos de pacientes del síndrome de Sanfilippo tipo C y un donante sano (Canals et al. 2015a). No obstante, para un correcto estudio de la enfermedad se debe tener en cuenta que el fondo genético de las distintas líneas celulares puede afectar a los resultados. Por este motivo, la generación de líneas isogénicas en las que la única diferencia entre la línea afectada y el control sea la alteración del gen causante de la enfermedad, se está convirtiendo en una estrategia muy utilizada. En los últimos años, el desarrollo de la tecnología de edición génica CRISPR/Cas9 ha supuesto un gran avance que ha facilitado enormemente la obtención de estas líneas isogénicas (Figura 1).
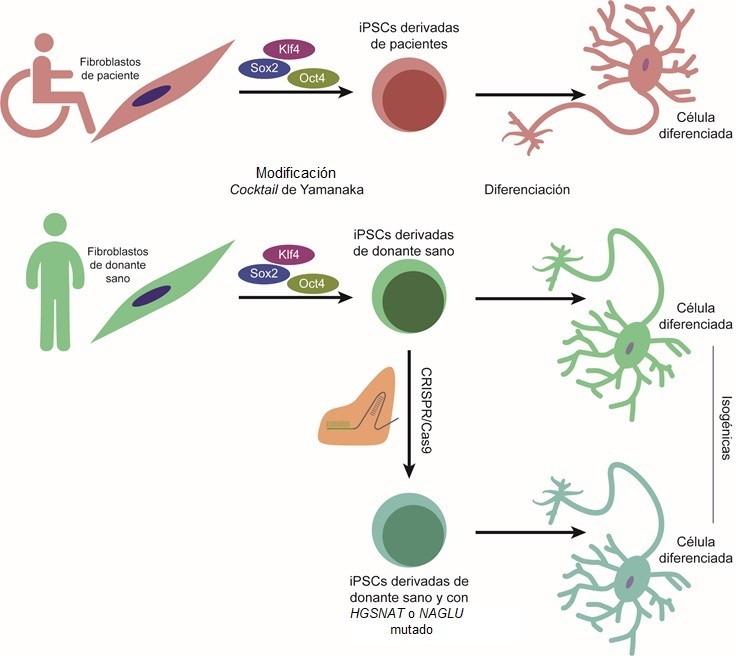
En un conjunto de trabajos hemos generado nuevos modelos celulares para el síndrome de Sanfilippo, siendo al mismo tiempo neurales y humanos. Además, los hemos usado para ensayar una aproximación terapéutica para esta enfermedad basada en el uso de siRNAs como terapia de reducción de sustrato.
En primer lugar, y usando la línea de iPSCs derivadas de fibroblastos de un donante sano (iPSCs WT) que se había obtenido previamente en nuestro laboratorio, se generaron y caracterizaron dos líneas isogénicas con mutaciones en el gen HGSNAT (causante del subtipo C de Sanfilippo) (Benetó et al, 2019), y otras dos en el gen NAGLU (subtipo B) (Benetó et al, 2020b) usando la tecnología de edición génica CRISPR/Cas9 en formato de ribonucleoproteína. En ambos casos se buscaba la alteración del gen mediante el mecanismo de unión de los extremos no homólogos (en inglés non-homologous end joining, NHEJ), una estrategia que suele ser más eficiente que la reparación homóloga (en inglés homology-directed repair, HDR) aunque más variable, dado que introduce inserciones o deleciones al azar en la región genómica de interés.
En el caso de HGSNAT, todas las mutaciones generadas causaban una alteración en la pauta de lectura provocando la aparición de un codón de parada prematuro. En cuanto a NAGLU, una de las líneas generadas también presentaba un codón de parada prematuro, pero en la otra encontramos una deleción de 9 pares de bases que daba como resultado la pérdida de 3 aminoácidos en la secuencia proteica. Sin embargo, en cualquiera de los casos, al comparar la actividad enzimática de las líneas mutadas con la WT, esta se reducía más del 80%, alcanzando valores muy similares a los detectados en iPSCs derivadas directamente de pacientes (Canals et al, 2015b). De esta forma se pudo confirmar la severidad de todas las mutaciones.
Posteriormente, se comprobó que todas las líneas de iPSCs obtenidas mantenían su estado pluripotente y conservaban su potencial de diferenciación a las tres capas embrionarias. El siguiente paso era diferenciarlas a tipos celulares neurales para poder así generar modelos celulares efectivos para el análisis de posibles tratamientos para el síndrome de Sanfilippo. Además, al tratarse de líneas isogénicas, los resultados obtenidos usando estos modelos son más robustos.
Para ello se usaron protocolos de diferenciación basados en la sobreexpresión de factores de transcripción para la obtención de neuronas y astrocitos (Benetó et al, 2020b), que permiten una diferenciación rápida y eficiente (Figura 1). Las líneas usadas fueron las dos con mutaciones en HGSNAT generadas con CRISPR/Cas9, la línea original iPSCs WT, y una de las derivadas de fibroblastos de paciente (Canals et al, 2015a).
A los 10 días de diferenciación se valoró la presencia de marcadores específicos de tipo celular para confirmar la correcta diferenciación de las iPSCs a los tipos celulares deseados y se decidió usar las neuronas obtenidas para realizar un examen más exhaustivo del fenotipo. Vimos que las líneas mutadas presentaban 3,5 veces más cantidad de HS que la WT. También se cuantificaron los lisosomas presentes en las distintas líneas, ya que un incremento de estos está asociado con la enfermedad, y se detectó una tendencia al alza en todas las líneas mutadas.
Nuestro grupo había descrito previamente el uso de siRNAs contra genes implicados en la síntesis del heparán sulfato como potencial terapia de reducción de sustrato para el síndrome de Sanfilippo (Canals et al, 2015a), usando fibroblastos de pacientes para ese análisis. Ahora queríamos utilizar la misma aproximación sobre los nuevos modelos neuronales de enfermedad para evaluar el efecto del siRNA más efectivo sobre fibroblastos (Benetó et al, 2020b) (Figura 2).
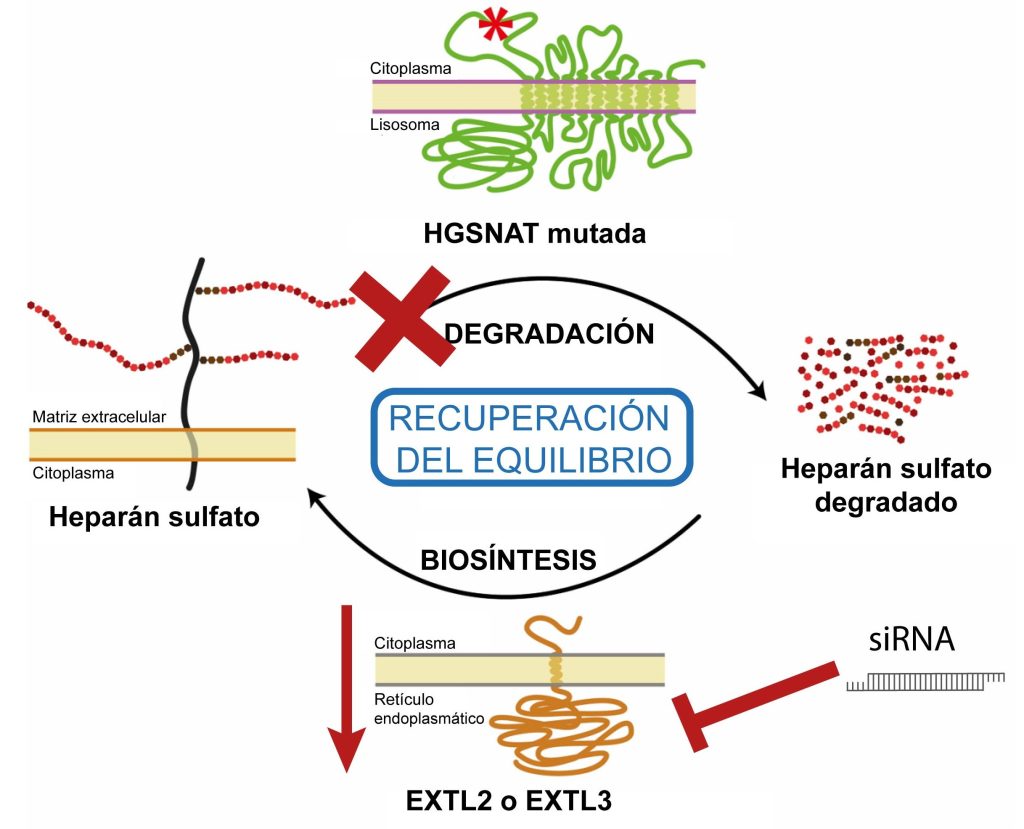
En primer lugar, mediante PCR cuantitativa en tiempo real se evaluó la eficiencia del siRNA, obteniendo una reducción en la expresión del mRNA del gen diana de alrededor del 75%. Sin embargo, a los 3 días de la transfección del siRNA en las neuronas, los niveles de HS no mostraron ninguna reducción.
Este es el primer estudio en el que se han generado nuevos modelos celulares neuronales humanos, de forma rápida y eficiente, resultando de gran utilidad para el ensayo de potenciales aproximaciones terapéuticas, como es en este caso una terapia de reducción de sustrato basada en el uso de siRNAs que se mostró eficiente en fibroblastos. Nuestros resultados demuestran la importancia de testar posibles terapias en líneas celulares de interés derivadas de los pacientes.

Artículos originales:
Benetó N, et al. Generation of two compound heterozygous HGSNAT-mutated lines from healthy induced pluripotent stem cells using CRISPR/Cas9 to model Sanfilippo C syndrome. Stem Cell Res. 2019 Dec;41:101616. doi: http://dx.doi.org/10.1016/j.scr.2019.101616.
Benetó N, et al. Generation of two NAGLU-mutated homozygous cell lines from healthy induced pluripotent stem cells using CRISPR/Cas9 to model Sanfilippo B syndrome. Stem Cell Res. 2020a Jan;42:101668. doi: http://dx.doi.org/10.1016/j.scr.2019.101668.
Benetó N, et al. Neuronal and Astrocytic Differentiation from Sanfilippo C Syndrome iPSCs for Disease Modeling and Drug Development. J Clin Med. 2020b Feb 28;9(3):644. doi: http://dx.doi.org/10.3390/jcm9030644.
Canals I, et al. EXTL2 and EXTL3 inhibition with siRNAs as a promising substrate reduction therapy for Sanfilippo C syndrome. Sci Rep. 2015a Sep 8;5:13654. doi: http://dx.doi.org/10.1038/srep13654.
Referencias:
Benetó N, et al. Sanfilippo Syndrome: Molecular Basis, Disease Models and Therapeutic Approaches. Int J Mol Sci. 2020c Oct 22;21(21):E7819. doi: 10.3390/ijms21217819.
Canals I, et al. Activity and High-Order Effective Connectivity Alterations in Sanfilippo C Patient-Specific Neuronal Networks. Stem Cell Reports. 2015b Oct 13;5(4):546-57. doi: 10.1016/j.stemcr.2015.08.016.
Si te ha gustado esta noticia y quieres aprender más sobre Genética en Medicina, te interesan nuestros cursos y formación universitaria.



