Un estudio recientemente publicado en The New England Journal of Medicine muestra resultados positivos para una terapia génica para la anemia falciforme basada en silenciar la expresión de un gen para aumentar los niveles de hemoglobina fetal en las células precursoras de los eritrocitos.
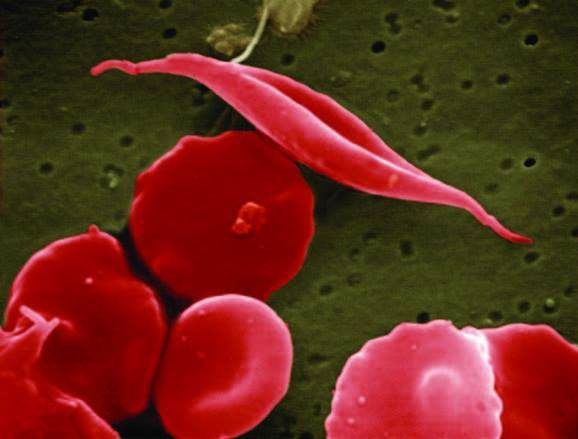
Investigadores de la Universidad de Harvard y el Instituto Dana Farber del Cáncer y Trastornos de la Sangre de Boston han tratado la anemia falciforme en seis pacientes a través de una terapia que recupera la expresión de la hemoglobina fetal en las células precursoras de la sangre. Con esta estrategia el equipo ha conseguido compensar la acción de la beta globina anómala que causa la enfermedad, así como reducir sus síntomas.
La anemia falciforme es una enfermedad genética producida por un cambio en el gen HBB, responsable producir la subunidad beta de la hemoglobina. La variante de la hemoglobina resultante, denominada HbS, provoca que los eritrocitos resultantes adquieran una forma anómala comprometiendo su función y ocasionando una serie de síntomas entre los que se encuentran la anemia hemolítica crónica y episodios dolorosos provocados por el flujo anómalo de eritrocitos en los vasos sanguíneos.
La terapia génica desarrollada por los investigadores tiene como objetivo evitar el silenciamiento de la proteína gamma globina fetal en los adultos. Durante el desarrollo embrionario y la infancia los niveles de expresión del gen HBB son bajos debido a que la hemoglobina predominante, conocida como hemoglobina fetal, no contiene proteína beta globina, sino gamma globina, producida por el gen HBG. Con la edad, la hemoglobina fetal es sustituida por la hemoglobina formada por dos subunidades de alfa globina y dos subunidades de beta globina, estas últimas producidas por el gen HBB. A partir de ese momento, es cuando se manifiesta de forma más acusada la presencia de alteraciones en el gen HBB que llevan a enfermedades como la anemia falciforme o beta talasemia.
Los pacientes con anemia falciforme muestran una versión mutante del gen HBB pero mantienen intacto el gen HBG de la hemoglobina fetal. Por esta razón, desde hace años se ha planteado la reactivación de la hemoglobina fetal como mecanismo para hacer frente a las enfermedades causadas por mutaciones en HBB.
La estrategia utilizada para recuperar la expresión de la beta hemoglobina fetal es actuar sobre otra proteína, BCL11A, que reprime su expresión a través de la unión a regiones reguladoras de HBG. El equipo ha tratado a seis pacientes con una terapia dirigida a inactivar BCL11A en las células madre precursoras de los eritrocitos. Esta aproximación específica de un tipo celular concreto evita comprometer las funciones del gen en otros tipos celulares.
La terapia consiste en extraer células madre hematopoyéticas del paciente, tratarlas para evitar que produzcan BCL11A, cultivarlas hasta que alcancen el número adecuado y volverlas a introducir en el paciente. Para modificar la expresión de BLC11A los investigadores utilizan un vector lentiviral que incluye un microARN bajo el control de elementos reguladores del gen HBB. El microARN está diseñado para unirse al ARN mensajero de BCL11A y suprimir su expresión de forma que se aumente la expresión de la beta globina fetal y se reduzca al mismo tiempo la expresión de la beta globina falciforme.
Los investigadores indican que las células modificadas fueron bien aceptadas por los pacientes. Además, tras un seguimiento de al menos seis meses, los pacientes mostraban una inducción estable de la expresión de hemoglobina fetal y habían experimentado una reducción significativa de los síntomas de la anemia falciforme. Respecto a los efectos adversos de la terapia, el equipo indica que son los habituales que se presentan en tratamientos similares.
Los resultados del trabajo ofrecen una vía terapéutica para enfermedades causadas por alteraciones en el gen HBB, como la anemia falciforme o la beta talasemia, causada por mutaciones que afectan a la producción de proteína beta globina. Además, representan un ejemplo de la aplicabilidad de terapias génicas dirigidas a reprimir la expresión de un gen.
A raíz de los resultados del trabajo los investigadores plantean que una estrategia alternativa podría ser la utilización del sistema CRISPR de edición del genoma para modificar la región intensificadora de la expresión de BCL11A específica del linaje de las células eritroides. Una aproximación similar está siendo investigada por la empresa CRISPR Therapeutics, que el año pasado anunció resultados prometedores en un paciente con anemia falciforme y un paciente con beta talasemia para una terapia que eliminaba la región intensificadora de BCL11A.
Artículo científico: Esrick EB, et al. Post-Transcriptional Genetic Silencing of BCL11A to Treat Sickle Cell Disease. NEJM. 2020. DOI: http://dx.doi.org/10.1056/NEJMoa2029392
Si te ha gustado esta noticia y quieres aprender más sobre Genética en Medicina, te interesan nuestros cursos y formación universitaria, así como nuestro canal audiovisual, Genotipia TV.