
Irantzu Pallarès y Salvador Ventura
Instituto de Biotecnología y Biomedicina. Universidad Autónoma de Barcelona
La agregación de proteínas en estructuras de tipo amiloide está asociada a la aparición de un número creciente de trastornos muy debilitantes o incluso fatales (Chiti y Dobson, 2006). La transtiretina (TTR) es una proteína homotetramérica cuya función fisiológica es el transporte de la hormona tiroidea T4 y y la proteína fijadora de retinol en el plasma. Es producida principalmente en el hígado y en los plexos coroideos del cerebro, y circula en plasma y en líquido cefalorraquídeo.
La amiloidosis por transtiretina (ATTR) es la forma más común de amiloidosis familiar en todo el mundo. En España, existen casos de esta enfermedad diseminados por todo el país, sin embargo, la prevalencia de casos es mayor en Palma de Mallorca, quinto foco mundial de la enfermedad. En la ATTR, mutaciones desestabilizantes en la TTR dan lugar a la formación de fibras amiloides que, dependiendo de la mutación, se depositan en diferentes órganos, tales como el cerebro, los nervios o el miocardio, provocando su malfuncionamiento, y dando lugar a las diversas formas de la enfermedad. La agregación de la TTR (ATTR) está asociada con la amilodosis sistémica senil (ASS) (Westermark et al., 1990), la miocardiopatía amiloide familiar (CAF) (Rapezzi et al., 2010) y la polineuropatía amiloide familiar (FAP) (Hou et al., 2007).
La ATTR se transmite como un rasgo autosómico dominante y se considera que la disociación del tetrámero de TTR en monómeros es el paso determinante de la patogenia. Hasta la fecha, se han descrito más de 100 mutaciones diferentes del gen de TTR (Johnson et al., 2012). Existe una forma leptomeníngea relativamente rara de la amiloidosis inducida por varias mutaciones puntuales que afecta al sistema nervioso central. La angiopatía amiloide cerebral y la amiloidosis ocular son las características clínicas comunes de este tipo de amiloidosis (Azebedo et al., 2011).
Durante muchos años, las opciones terapéuticas para paliar el progreso de la enfermedad en los pacientes con ATTR han sido el trasplante de hígado o el trasplante combinado de hígado y corazón, con la intención de eliminar casi por completo la producción de la variante anómala de la proteína y detener así la progresión de la enfermedad (Herlenius, et al., 2004). Sin embargo, el trasplante no siempre impide eficazmente el avance de la enfermedad y no procede como tratamiento de la forma leptomeníngea.
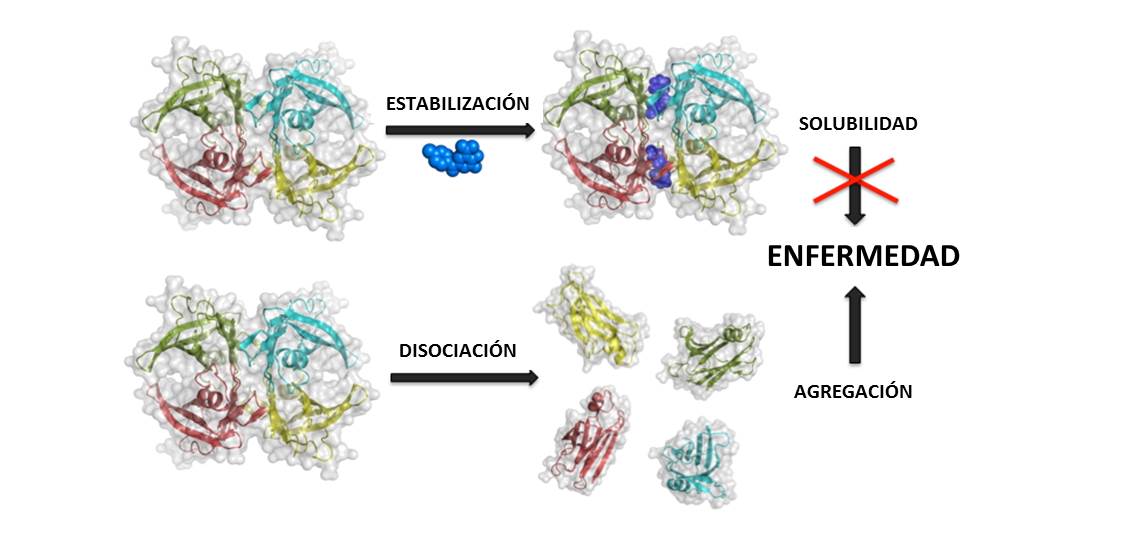
El uso de agentes estabilizantes de la estructura de TTR está surgiendo como un nuevo método terapéutico no invasivo para detener el curso de la enfermedad. En esta línea de trabajo, nuestro grupo, en una colaboración con la empresa biofarmacéutica SOM Biotech, ha descrito recientemente cómo la aplicación de una estrategia de reposicionamiento de fármacos para la enfermedad de la ATTR ha culminado en el descubrimiento de tolcapone como un potente fármaco que puede mejorar significativamente el tratamiento farmacológico de la ATTR (Sant’Anna et al., 2016). SOM Biotech ha protegido ya mediante una patente el uso de de esta molecula para el tratamiento de la ATTR. Se trata de una propiedad hasta ahora desconocida de este fármaco, que ya se utiliza para el tratamiento del Párkinson.
Los datos moleculares y estructurales obtenidos en nuestro trabajo, demuestran que tolcapone actúa imitando el proceso de unión de la hormona tiroidea T4 con la TTR, al ser transportada por la sangre. Igual que la hormona, el fármaco se une fuertemente a la proteína, aumentando la barrera energética de disociación del tetrámero en monómeros. Los ensayos biofísicos, in vitro en cultivos celulares, ex vivo en plasma de personas y en ratones modelos de la enfermedad constataron que tolcapone es un potente inhibidor del inicio del proceso de agregación de fibras amiloides por TTR, que actúa estabilizando la estructura de la proteína, lo que reduce la progresión de la enfermedad. El compuesto se ha mostrado hasta cuatro veces más eficaz que el único medicamento que hay actualmente para tratar la variante polineuropática de la enfermedad (Berk et al., 2013). Los resultados han sido positivos para todas las variantes de la enfermedad estudiadas: la polineuropatía y la cardiomiopatía amiloide familiar (que afectan a los nervios periféricos y al miocardio, respectivamente) y la amiloidosis sistémica senil, una forma esporádica que afecta el miocardio de un porcentaje muy elevado de personas mayores de 60 años. Además, puesto que es capaz de atravesar la barrera hematoencefálica podría suponer el primer tratamiento para las variantes amiloidogénicas de TTR que afectan al sistema nervioso central. Tolcapone tiene el potencial para convertirse en una molécula eficaz para prevenir las deposiciones de la proteína que causan la enfermedad y retardar su progresión, y podría estar en el mercado en un plazo relativamente corto, debido a que ya se ha probado en un ensayo clínico con personas afectadas por la variante neuropática.
El reposicionamiento de fármacos permite identificar la idoneidad de moléculas ya aprobadas para una determinada indicación terapéutica -como es el caso del tolcapone para el tratamiento del Párkinson- para la terapéutica de una enfermedad diferente, acelerando así el desarrollo y el acceso a los pacientes a nuevos tratamientos. Esta estrategia también favorece un menor coste de los tratamientos, lo que podría facilitar, en el caso del tolcapone, la administración en países como Brasil o Portugal, importantes focos de la variante polineuropática. El medicamento ha recibido la designación de medicamento huérfano para la ATTR por parte de la Food and Drug Administration norteamericana. Un hecho relevante, puesto que en Estados Unidos hay un grupo importante de población afectada por la variante cardiomiopática de la enfermedad.
Referencia: Sant’Anna R, et al. Repositioning tolcapone as a potent inhibitor of transthyretin amyloidogenesis and associated cellular toxicity. Nat Commun. 2016. 7, 10787. doi: http://dx.doi.org/10.1038/ncomms10787
Bibliografía:
Chiti, F. y Dobson, C M. Protein misfolding, functional amyloid, and human disease. Annual rev biochem. 2006. 75, 333-366. doi:10.1146/annurev.biochem.75.101304.123901
Westermark P, et al. Fibril in senile systemic amyloidosis is derived from normal transthyretin. Proc Natl Acad Sci U S A. 1990. 87, 2843-2845.
Rapezzi, C, et al. Transthyretin-related amyloidoses and the heart: a clinical overview. Nat Rev Cardio. 2010. 7, 398-408. doi:10.1038/nrcardio.2010.67
Hou, X, et al. Transthyretin and familial amyloidotic polyneuropathy. Recent progress in understanding the molecular mechanism of neurodegeneration. FEBS J. 2007. 274, 1637-1650.
Johnson SM, et al. The transthyretin amyloidoses: from delineating the molecular mechanism of aggregation linked to pathology to a regulatory-agency-approved drug. J Mol Biol. 2012. 421, 185-203. doi: 10.1016/j.jmb.2011.12.060.
Azevedo EP et al. Dissecting the structure, thermodynamic stability, and aggregation properties of the A25T transthyretin (A25T-TTR) variant involved in leptomeningeal amyloidosis: identifying protein partners that co-aggregate during A25T-TTR fibrillogenesis in cerebrospinal fluid. Biochemistry. 2011. 50, 11070-11083. doi: 10.1021/bi201365r.
Herlenius G, et al. Ten years of international experience with liver transplantation for familial amyloidotic polyneuropathy: results from the Familial Amyloidotic Polyneuropathy World Transplant Registry. Transplantation. 77, 64-71.
Berk JL, et al. Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. JAMA. 2013. 310, 2658-2667. doi: 10.1001/jama.2013.283815.

