Las técnicas de secuenciación de fragmentos largos del genoma permiten identificar una expansión de ADN en el gen ZFHX3 como causa genética de la ataxia espinocerebelosa tipo 4, abriendo la posibilidad de diagnóstico genético para esta enfermedad rara, así como el desarrollo de estrategias terapéuticas.
La ataxia espinocerebelosa de tipo cuatro (también llamada SCA4, por sus siglas en inglés) es una enfermedad neurológica poco frecuente caracterizada por ataxia y pérdida sensorial periférica de aparición tardía. Su carácter hereditario se conoce desde hace tiempo. Como otras ataxias, se transmite con un patrón de herencia autosómico dominante, lo que indica que una única copia afectada del genoma es suficiente para el desarrollo de la enfermedad. Sin embargo, la causa genética exacta, es decir, qué alteración y qué genes están implicados, continuaba siendo una incógnita hasta hace muy poco.
En 1996, a través del análisis de cinco generaciones de una única familia de Utah, EE.UU., se había podido acotar que la causa de la enfermedad se encontraba en una región del cromosoma 6. No obstante, no fue posible identificar el cambio genético concreto responsable de la enfermedad, debido a la dificultad de analizar la región implicada, rica en nucleótidos C y G, así como en duplicaciones y pseudogenes.
Utilizando las últimas tecnologías de secuenciación un equipo internacional, liderado por Stefan Pulst y Pattie Figueroa, de la Universidad de Utah, ha conseguido identificar una expansión de ADN en el gen ZFHX3 como responsable de la SCA4. Además, los investigadores han detectado cinco pacientes más portadores de la alteración que habían sido diagnosticados con ataxia pero no tenían un diagnóstico más preciso. Los resultados se han publicado en la revista Nature Genetics.
Una expansión del gen ZFHX3 como causa de la ataxia espinocerebelosa tipo 4
Para identificar la causa genética de la SCA4, los investigadores recurrieron a la tecnología de secuenciación de Pacific Biosciences y la secuenciación mediante nanoporos de Oxford Nanopore Technologies. Estas aproximaciones permiten secuenciar fragmentos largos de ADN lo que facilita el análisis de regiones con repeticiones que incluyen nucleótidos C y G, o regiones ricas en fragmentos que pueden encontrarse repetidos en otras partes del genoma.
Al comparar los genomas miembros afectados y no afectados de la familia de Utah los investigadores detectaron una variante no identificada hasta el momento que reunía las características para ser considerada una posible causa de la SCA4: estaba presente únicamente en los familiares afectados, se trataba de una expansión de un trinucleótido (algo frecuente en otras ataxias), era muy poco frecuente y fue detectada en cinco pacientes independientes a la familia que tenían ataxia y neuropatía sensorial sin causa conocida hasta el momento.
La variante en cuestión consiste en una expansión del trinucleótido CGG en el gen ZFHX3. La versión más frecuente del gen tiene 21 repeticiones, mientras que en los pacientes con SCA4, este número se extiende a entre 42 y 74 repeticiones.
ZFHX3 codifica para una proteína que actúa como factor de transcripción, regulando la expresión de múltiples genes. Hasta el momento se sabía que la pérdida de función del gen lleva a un trastorno del neurodesarrollo que implica discapacidad intelectual y dismorfología facial, un fenotipo muy diferente al observado en pacientes con SCA4, que no muestran características de neurodesarrollo.
La expansión de trinucleótidos detectada en los pacientes con SCA4 provoca que una región de la proteína ZFHX3, formada por repeticiones del aminoácido glutamina, sea más larga de lo normal. A partir del análisis de cerebros post mortem de pacientes y estudios funcionales en células los investigadores plantean que el efecto patológico podría estar relacionado con el procesamiento de proteínas mal plegadas en la célula y alteraciones en la autofagia. “Esta mutación es una repetición expandida tóxica y creemos que bloquea la forma en que la célula gestiona las proteínas desplegadas o mal plegadas”, ha destacado Pulst.
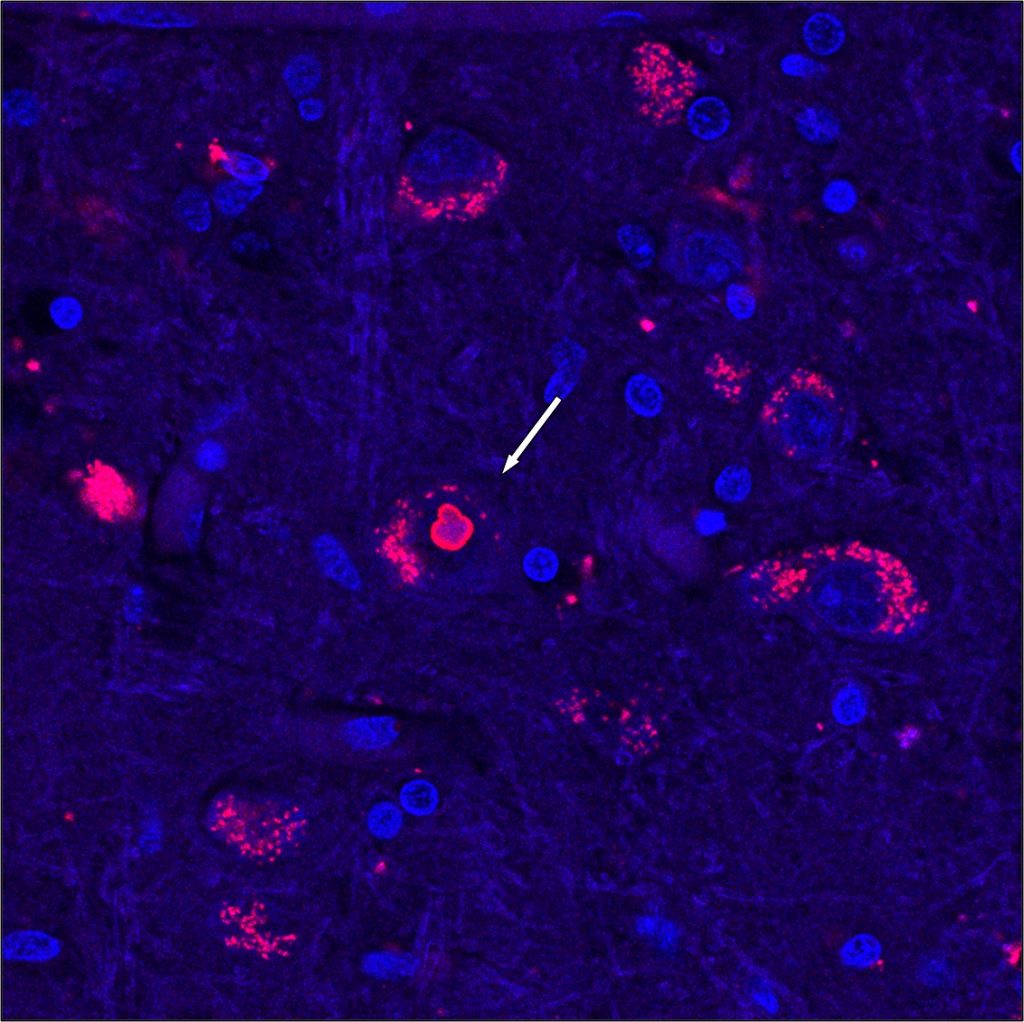
Implicaciones de la identificación de la expansión CGG como causa de SCA4
La identificación de la mutación genética responsable de la SCA4, que culmina una investigación iniciada hace décadas, tiene impacto sobre el diagnóstico y tratamiento de la enfermedad.
En primer lugar, facilita el diagnóstico de pacientes y abre la posibilidad a que los familiares de personas afectadas puedan realizarse pruebas genéticas para determinar si son portadores de la mutación. Aunque se trata de una enfermedad de aparición tardía, conocer el riesgo puede contribuir a la hora de tomar decisiones vitales y, quizás en el futuro, permita también establecer medidas preventivas.
Por otra parte, los resultados plantean algunas opciones terapéuticas para la enfermedad, que en estos momentos no tiene cura. Al pasar a formar parte de las enfermedades producidas por expansiones de poliglutaminas, los investigadores estiman que podría beneficiarse de terapias para este tipo de enfermedades que ya están en desarrollo o fase de ensayos clínicos en la actualidad.
Artículo científico: Figueroa, K.P., Gross, C., Buena-Atienza, E. et al. A GGC-repeat expansion in ZFHX3 encoding polyglycine causes spinocerebellar ataxia type 4 and impairs autophagy. Nat Genet (2024). https://doi.org/10.1038/s41588-024-01719-5
Fuente: After 25 years, researchers uncover genetic cause of rare neurological disease. https://healthcare.utah.edu/newsroom/news/2024/04/after-25-years-researchers-uncover-genetic-cause-of-rare-neurological-disease
Si te ha gustado esta noticia y quieres aprender más sobre Genética en Medicina, te interesan nuestros cursos y formación universitaria, como el “Experto Universitario en Neurogenética” así como nuestro canal audiovisual, Genotipia TV.



