Ana María García Marín, Universitat de València; Álvaro Chiner Oms, Instituto de Biomedicina de Valencia (IBV – CSIC); Fernando González Candelas, Universitat de València; Iñaki Comas Espadas, Instituto de Biomedicina de Valencia (IBV – CSIC); Mariana Gabriela López, Instituto de Biomedicina de Valencia (IBV – CSIC) y Mireia Coscolla Devis, Universitat de València
Durante los últimos diez años el mundo de la genómica de patógenos ha experimentado una notable transformación. La confluencia de tres factores ha sido determinante para alcanzar su auge actual.
En primer lugar, tenemos la capacidad de secuenciar miles de genomas de un patógeno a un precio asequible. En segundo lugar, gracias a herramientas prestadas de la filogenética y la evolución molecular, podemos establecer las relaciones genéticas entre dichas muestras a una escala poblacional antes impensable. Por último, tenemos la capacidad de secuenciar a tiempo casi real.
Esto nos permite integrar la información genómica con la epidemiológica, para transformar las relaciones entre genomas en patrones de dispersión de una enfermedad. Como resultado, podemos desarrollar estudios de epidemiología genómica con un impacto directo en las actuaciones de salud pública.
La aparición del SARS-CoV-2 ha propiciado la aplicación de la epidemiología genómica en una respuesta sin precedentes a nivel internacional. En España, los autores de este artículo representamos al consorcio SeqCOVID, coordinado por el CSIC y financiado en su mayor parte por el Fondo COVID del Instituto de Salud Carlos III. El consorcio está compuesto por más de 50 grupos de investigación y hospitales nacionales.
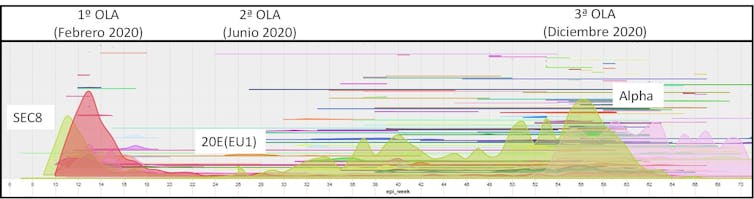
Esta iniciativa ha logrado que nuestro país sea uno de los principales contribuidores de secuencias de SARS-CoV-2 del mundo al inicio de la pandemia. Este esfuerzo nos ha permitido estudiar la evolución del virus, así como la aparición y desaparición de variantes en España desde Febrero de 2020. De hecho, si bien unas pocas variantes atraen la atención, tanto en España como en el mundo ha habido miles de ellas (Figura 1). A continuación relataremos lo que hemos aprendido de la pandemia desde una visión de la genómica del virus.
Primera ola: ¿Cómo entró el virus al país?
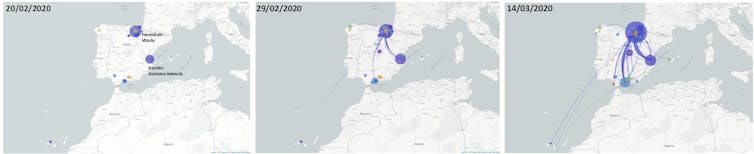
El primer reto del consorcio fue desentrañar cómo comenzó la pandemia en España. Inicialmente, se analizó el 14% de los casos antes del confinamiento y el 1% de la primera ola. El análisis comparativo con muestras de todo el mundo reveló la inexistencia de un ‘paciente 0’. Es decir, el coronavirus no se dispersó por el país a partir de un único paciente.
En contraposición, la diversidad genómica del virus era alta. En un contexto internacional, los virus circulantes en España se parecían mucho a los circulantes en un gran número de países. Sin embargo, al contrario que en otros países europeos, existía una alta representación de los linajes A.
Dichos linajes fueron los primeros en circular en Asia, y particularmente en China. Esto no significa que el virus fuera importado únicamente desde el país asiático. Nuestros análisis permitieron identificar un gran número de fuentes iniciales. Se definieron hasta un total de 519 introducciones diferentes del virus en todo el territorio nacional.
Esta cifra debe ser tomada con cautela, ya que el número de secuencias analizadas fue mucho menor que el de casos confirmados. Aún así, es igualmente indicativa del gran número de importaciones independientes que, con mayor o menor éxito, contribuyeron a la epidemia inicial.
A su vez, las fechas señalan que las exposiciones se aceleraron a partir del 24 de febrero, manteniéndose constantes hasta casi el estado de alarma. El estudio también reveló la existencia de múltiples grupos de transmisión compuestos por más de cinco individuos. Esto concuerda con el modo de transmisión del SARS-CoV-2 en eventos de superdispersión.
Unas pocas introducciones tuvieron un gran éxito
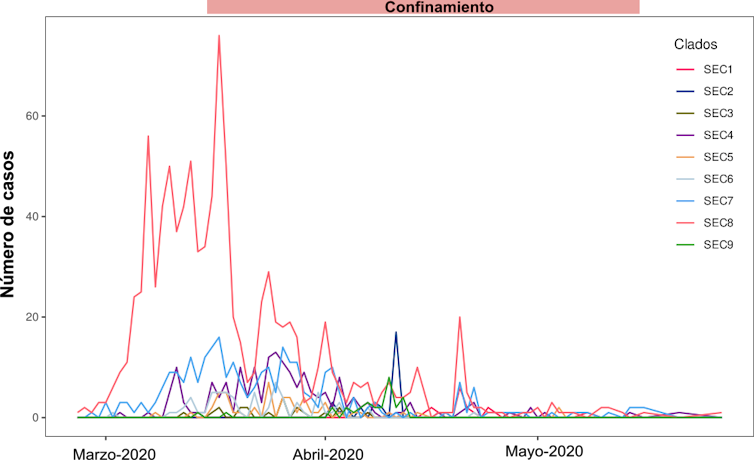
El análisis de la filogenia de las muestras secuenciadas mostraba que no agrupaban todas juntas. En él se diferenciaban grandes grupos filogenéticos enriquecidos en muestras españolas. Esto sugería que eran variantes expandidas principalmente por el territorio nacional y no por otras partes del mundo. Se identificaron así nueve grupos que denominamos ‘Clados Epidémicos Españoles’ o SEC (por sus siglas en inglés)(Figura 2).
Ellos son los responsables de la mayoría de los casos del periodo inicial. Especialmente los SEC7 y SEC8 englobaban un gran número de secuencias de la primera ola. De hecho, representaban el 10% y el 30% de todas las muestras españolas respectivamente. También fueron particularmente prevalentes antes del confinamiento. El 60% de los casos de la semana previa al inicio del estado de alarma pertenecían al SEC8.
Para cada grupo se identificaron las variantes ancestrales de las que derivaron el resto de casos. En los SEC7 y SEC8 estas variantes estaban relacionadas con las variantes que aparecieron originalmente en China. Sin embargo, en el resto de Europa predominaron variantes más tardías. Esto nos indica, con alta probabilidad, que en nuestro país se produjeron muy pronto las primeras importaciones del virus.
Además, estos dos clados se dispersaron ampliamente en poco tiempo por la geografía española. La distancia geográfica media entre sus muestras era superior al resto. Concluimos así que los SEC7 y SEC8 fueron los dos grupos más exitosos. Y por tanto, también los responsables del desarrollo inicial de la epidemia en España.
¿Cuándo se produjeron las primeras introducciones del virus?
Para responder a esta pregunta se analizó la acumulación de cambios genéticos del virus a lo largo del tiempo. Nuestras estimaciones sugieren que las introducciones más tempranas y exitosas del virus ocurrieron en febrero. Estas además se corresponden con casos de los grupos 7 y 8. También se corrobora que durante la segunda quincena de dicho mes el virus se expandió rápidamente.
Este período de introducción coincide temporalmente con el primer paciente diagnosticado en España. No obstante, no puede descartarse que el virus hubiera ingresado antes en el país. El muestreo al inicio de la pandemia fue reducido, y nuestra datación se corresponde con la evidencia epidemiológica disponible.
Aún así, si hubo introducciones previas, no ha quedado rastro posterior de las mismas en la epidemia. El origen de los otros SECs se sitúa entre finales del mes de febrero y principios del mes de marzo. En la mayor parte de los casos representan brotes grandes pero locales.
Éxito del SEC8
Saber exactamente a qué se debe el éxito de unas variantes y no de otras siempre es complejo. Al comienzo de una epidemia rara vez una mutación está asociada a éxito epidemiológico o virulencia. Son más probables los eventos fortuitos, particularmente eventos fundadores. Esto es lo que creemos que ocurrió con el SEC8, el grupo que mostró una dispersión especialmente rápida y exitosa por todo el país.
Nuestros análisis demuestran que parte de su éxito se explica por su pronta introducción en el país, previa a las medidas de restricción. A esta primera fase de introducciones tempranas le siguió una fase de incremento de la prevalencia hasta representar más del 60% de casos antes del confinamiento. Esta amplificación del número de casos estuvo ligada a eventos de superdispersión.
Por ejemplo, el SEC8 entró al menos dos veces desde Italia hasta Valencia como consecuencia del partido Atalanta-Valencia de la Champions League. En una de ellas ocasionó un número elevado de casos secundarios.
También se identificaron focos tempranos en Madrid, Andalucía y el País Vasco. De especial relevancia fue el evento de superdispersión originado por el famoso funeral de Vitoria, que reunió a asistentes del País Vasco y La Rioja junto a personas provenientes de países europeos. Finalmente, la ausencia de medidas de restricción de la movilidad motivó una rápida dispersión al resto del país (Figura 3). Como veremos más adelante, el impacto negativo de la alta movilidad en la evolución epidemiológica y en la redistribución de variantes es una constante en toda la pandemia.
¿Fueron útiles las medidas de confinamiento para frenar la expansión del virus?
El confinamiento es una medida con efectos drásticos a nivel social y económico. Por ello es imprescindible evaluar su impacto real en el control de la epidemia.
Para conocer la tasa de crecimiento de la pandemia antes y después del confinamiento, se calculó el número de reproducción efectiva o Re para los grupos SEC7 y SEC8. Este valor representa el número de infecciones secundarias que causa cada individuo infectado en una población determinada. Por tanto, las fluctuaciones de Re en el tiempo nos informan directamente en qué fase se encuentra la epidemia: crecimiento, decrecimiento o estabilización.
Según nuestras estimaciones, en el periodo previo al confinamiento el valor de Re para SEC7 y SEC8 se situaba alrededor de 2-3. Es decir, cada paciente infectado contagiaba de media a otras dos o tres personas. Este resultado concuerda con el valor de Re estimado para el SARS-CoV-2 con otros modelos epidemiológicos.
Al final de la primera ola en España, el Re disminuyó drásticamente en ambos grupos hasta el 0.27. Pasamos, así, a fase de decrecimiento. Para el SEC7 la caída empezó entre el 15 y 24 de marzo, justo tras la fecha de confinamiento. En el caso del SEC8, se produjo ligeramente antes, entre el 8 y 10 de marzo. Coincide con la implementación de las primeras restricciones parciales.
Llegamos así a la conclusión de que el confinamiento fue una medida altamente eficaz, que no solo contribuyó a reducir el número de casos y de fallecimientos sino a eliminar muchas de las variantes circulantes.
Segunda ola: una variante identificada en España se expande por Europa
Gracias al éxito del confinamiento, en la segunda ola cambiaron las variantes en circulación. Entre las muestras de esta segunda fase el rastro de los SECs es escaso y se detectan nuevas variantes. Destaca la variante denominada 20E (EU1) o B.1.177, que presenta una mutación adicional en la proteína S.
La voz de alarma surge tras detectarse simultáneamente por primera vez en un brote local en Castellón y en Suiza, donde crece muy rápidamente. Su origen se identifica en Aragón a finales de junio de 2020. Se trata de muestras asociadas a un gran evento de transmisión entre trabajadores de la fruta.
Los brotes en este tipo de trabajadores han sido comunes durante la epidemia. Se debe a su situación de vulnerabilidad, con condiciones de trabajo y vida precarias. Finalmente, también se detecta en un único caso en Holanda, lo que complica conocer su procedencia exacta.
En el periodo de postconfinamiento, con la circulación del virus reducida a mínimos, la nueva variante encuentra la oportunidad de ocupar un nicho vacío. Gracias a los eventos de superdispersión locales, se propició su rápida distribución por todo el territorio. Pasó, así, a ser la variante dominante durante el verano y el otoño.
En agosto, el 80% de las muestras secuenciadas ya pertenecían a esta variante. Esta proporción se mantuvo estable hasta enero de 2021. De nuevo, una variante del virus incrementa su prevalencia gracias a eventos de superdispersión y por la alta movilidad.
Queda por tanto, en evidencia, la falta de medidas de contención adecuadas para el control de la epidemia. Así como la importancia de proteger a las poblaciones más vulnerables.
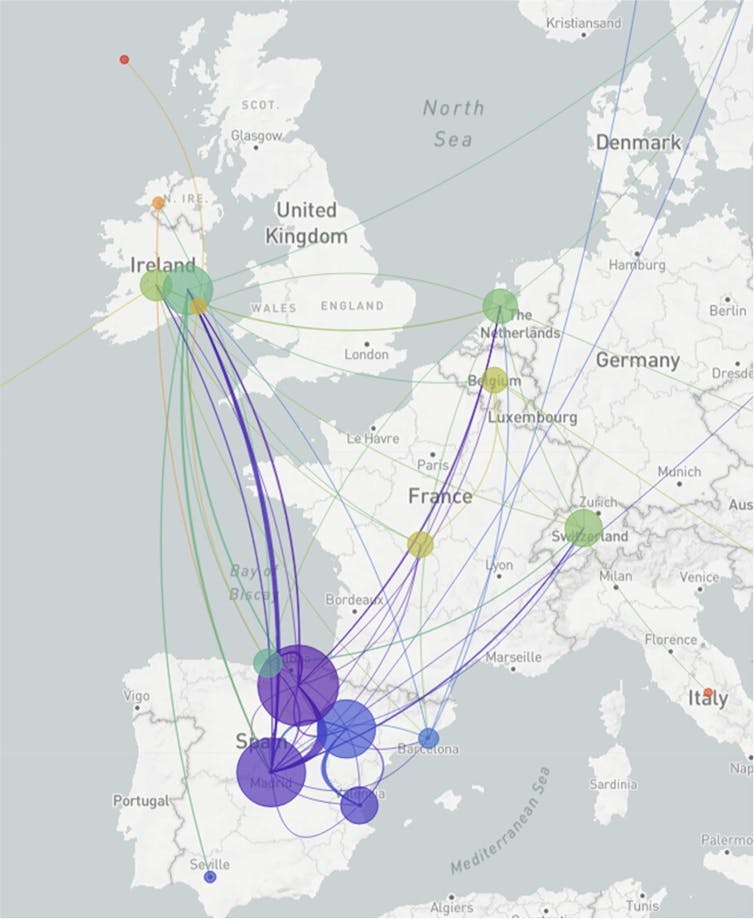
Este mismo patrón se vio ampliado a nivel de toda Europa. A mediados de junio, España abrió sus fronteras para el turismo. Esto posibilitó que muchos turistas se llevaran la variante a sus países de origen, favoreciendo su expansión por todo el continente (Figura 4).
Concretamente, en Reino Unido esta variante pasó a ser la dominante alrededor de octubre 2020. Paralelamente, se produjeron exportaciones e importaciones de dicha variante entre los diferentes países europeos. Para mediados de octubre ya había llegado a Asia. Incluso se produjo una importación en Nueva Zelanda a través de un brote sucedido en un avión que aterrizó en ese país.
Esto demuestra el pobre control de la transmisión que se hizo en Europa durante el verano y principios de otoño. Una situación internacional que es claro ejemplo de la aplicación de medidas de control de transmisión poco efectivas.
Tercera ola: ¿cómo afectó la aparición de la variante Alpha en España?
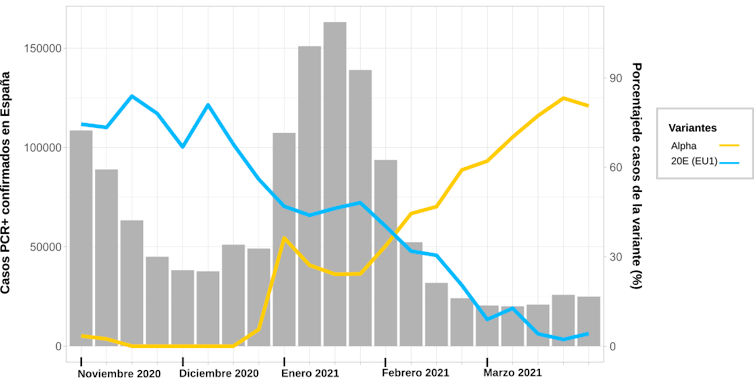
La tercera ola en España se ha caracterizado por la presencia de las mismas variantes que la segunda ola (Figura 5). En concreto, la 20E (EU1) ha sido la más común. Por tanto, este tercer periodo no derivó de la presencia de nuevas variantes más transmisibles. Fue consecuencia directa de las medidas relajadas que se adoptaron durante el periodo navideño.
No obstante, a finales de diciembre de 2020 el Reino Unido notificó la aparición de una nueva variante (finalmente denominada Alpha). En cuestión de semanas había desplazado al resto, sobre todo en el sudeste del país. Los primeros datos indicaban que en este caso sí era más transmisible. También sugerían que estaba detrás de gran parte de los casos observados durante la tercera ola del país anglosajón.
Posteriormente, se vio confirmado al mostrar trayectorias similares en otros países. La variante fue introducida en lugares como Irlanda, Portugal, Dinamarca o Suiza, donde dominó en la tercera ola. Estudios han demostrado que la variante Alpha efectivamente tiene mayor transmisibilidad probablemente ligada a una acumulación de mutaciones en la espícula que le confiere una ventaja evolutiva. Por ello, ha sido capaz de desplazar al resto de variantes en un escenario de alta competición y circulación del virus.
En España, la presencia de Alpha a finales de año era todavía limitada. La variante de verano seguía dominando en porcentaje de casos. Por tanto, no estuvo detrás de la tercera ola del país. Un tiempo después sí se observó un rápido crecimiento, al igual que en otros países europeos. Durante los primeros meses de 2021 se invirtieron las proporciones en nuestro país. La variante Alpha ganó la competición frente a la variante de verano, que quedó desplazada. Para el mes de abril, aproximadamente un 85% de los casos a nivel nacional ya se debían a la nueva variante (Figura 5). Sin embargo, en España, al contrario que en el Reino Unido, no generó una ola de casos tan acusada.
Por tanto, la cuarta ola asociada a Alpha en España nunca ha llegado a materializarse completamente. La razón es que el destino de una variante no está escrito. Su capacidad de poner en estrés al sistema depende tanto de la transmisibilidad de la variante como de nuestra capacidad para, no frenar, si no modular su avance. Y esta es una lección que se puede aplicar a cualquier otra variante.
Variantes de preocupación un año después de la pandemia
Durante la primera ola ya se empezaron a describir diferentes variantes de SARS-CoV-2, muchas de ellas con nulo impacto en la epidemia. De todas las mutaciones observadas, la mutación D614G en la espícula es la que podría estar asociada a una mayor transmisibilidad. Aún así, el efecto no es tan pronunciado como el observado en alguna de las variantes más recientes.
Ha hecho falta un año para empezar a detectar variantes que sí cambian las características del coronavirus con respecto a las iniciales. Sin embargo, hay que recalcar que un cambio en alguna característica no debe llevarnos al alarmismo. Muchas de esas variantes no han conseguido establecerse en nuestro país y las que lo han hecho, como la Alpha, se han controlado razonablemente bien.
Cuando hablamos de variantes debemos preguntarnos tres cosas:
- ¿Cambian sustancialmente la capacidad de transmisión del virus?
- ¿Están asociadas a mayor severidad de la enfermedad?
- ¿Son capaces de reducir la efectividad de las vacunas o de la respuesta inmune en general?
Las variantes que de momento se están imponiendo, sobretodo, son aquellas con una ventaja en la transmisión (Alpha, Delta). Hay muy poca evidencia de que Alpha, Delta o alguna otra provoque una enfermedad esencialmente más grave (algunos estudios sugieren que Alpha la puede ocasionar marginalmente).
Esto tiene un gran sentido evolutivo. La selección natural se refleja en la descendencia y, por tanto, es cuando se transmite el virus cuando puede actuar. En el caso de SARS-CoV-2, la transmisión se produce antes de los síntomas, o incluso en estado asintomático. Por tanto, la transmisión no está ligada a la severidad de la enfermedad. Dicho de otra manera, para cuando un paciente enferma de verdad el virus ya se ha transmitido y ha dejado descendencia, por lo que en realidad no le importa lo que le pase al paciente (para bien o para mal).
Respecto a variaciones en la interacción con el sistema inmune, sí existe evidencia de variantes que reducen la eficacia de anticuerpos neutralizantes. Es el caso de Beta, por ejemplo. Son variantes muy marginales en España. En cualquier caso, las vacunas mantienen la efectividad contra todas la variantes circulantes, incluyendo la Delta.
Mirando al futuro
Por primera vez en la pandemia existe un desacople entre la curva de casos y la de hospitalizaciones. Esto es gracias a la vacunación. Pero en España todavía es incompleta y no nos ayuda a parar los contagios actuales, sino a parar olas futuras. Por tanto, todavía debemos convivir con medidas de prevención de la transmisión.
Cada vez que el virus se transmite tiene una nueva oportunidad. Bajar la transmisión del virus hace que el sistema sanitario se mantenga a salvo de la saturación. A la vez, se mantienen bajo control las variantes existentes y se evita la aparición de otras nuevas. Esto cobra especial importancia en el contexto de poblaciones parcialmente o totalmente inmunizadas, donde la inmunización representa una nueva presión de selección para el virus.
En España, la variante Delta, identificada por primera vez en la India, está empezando a imponerse y es de prever que termine reemplazando a Alpha. La velocidad a lo que lo haga, esta o cualquier otra variante, y la magnitud de la ola a la que pueda estar asociada, dependen esencialmente de las trabas que pongamos entre todos a la transmisión de SARS-CoV-2. No repitamos el verano de 2020.
![]()
Ana María García Marín, Investigadora predoctoral en Unidad de Genómica de la Tuberculosis. (Instituto de Biomedicina de Valencia IBV-CSIC). Unidad mixta «Infección y Salud Pública» (FISABIO-UV), Universitat de València; Álvaro Chiner Oms, Investigador post-doctoral en la Unidad de Genómica de la Tuberculosis, Instituto de Biomedicina de Valencia (IBV – CSIC); Fernando González Candelas, Catedrático de Genética. Responsable Unidad Mixta de Investigación «Infección y Salud Pública» FISABIO-Universitat de València I2SysBio. CIBER Epidemiología y Salud Publica, Universitat de València; Iñaki Comas Espadas, Científico titular, Unidad de Genómica de la Tuberculosis, Instituto de Biomedicina de Valencia (IBV – CSIC); Mariana Gabriela López, , Instituto de Biomedicina de Valencia (IBV – CSIC) y Mireia Coscolla Devis, , Universitat de València
Este artículo fue publicado originalmente en The Conversation. Lea el original.
Si te ha gustado esta noticia y quieres aprender más sobre Genética en Medicina, te interesan nuestros cursos y formación universitaria, así como nuestro canal audiovisual, Genotipia TV.


