Autores: Víctor López del Amo, Marta Seco-Cervera, José Luís García-Giménez, Alexander J. Whitworth, Federico V. Pallardó y Máximo Ibo Galindo
La enfermedad de Charcot-Marie-Tooth (CMT) es una enfermedad neurodegenerativa que por su índice de prevalencia (1 de cada 2500 personas aproximadamente) está englobada dentro del grupo de enfermedades raras, aún así es una de las enfermedades más representativas dentro de dicho grupo. Se trata de una enfermedad muy heterogénea desde el punto de vista clínico y genético pero con un punto en común, todas afectan al sistema nervioso periférico. Los pacientes presentan una sintomatología desde leve hasta muy grave y hay más de 50 genes causantes de CMT, tanto de herencia dominante como recesiva.
Clásicamente la clasificación distingue dos grupos, según electrofisiología e histopatología:
– CMT1 o formas desmielinizantes
– CMT2 o formas axonales
Uno de los genes que provoca la enfermedad de CMT es el gen GDAP1, y codifica para una proteína mitocondrial localizada en la membrana externa que participa en los procesos de dinámica mitocondrial. Desde 2002, cuando se relacionó pacientes con mutaciones en GDAP1 y la enfermedad de CMT, se ha avanzado mucho en la comprensión de los mecanismos moleculares de la proteína en particular, pero no se ha estudiado el efecto de la proteína en un entorno más complejo como es un organismo completo.
Por ello, en un trabajo dirigido por el Dr. Máximo Ibo Galindo y en colaboración con la Facultad de Medicina de la Universitat de València y la Universidad de Sheffield, se ha creado un nuevo modelo en Drosophila que permite simular los defectos acontecidos en pacientes y de esta manera estudiar en profundidad cuales son los mecanismos que producen la enfermedad.
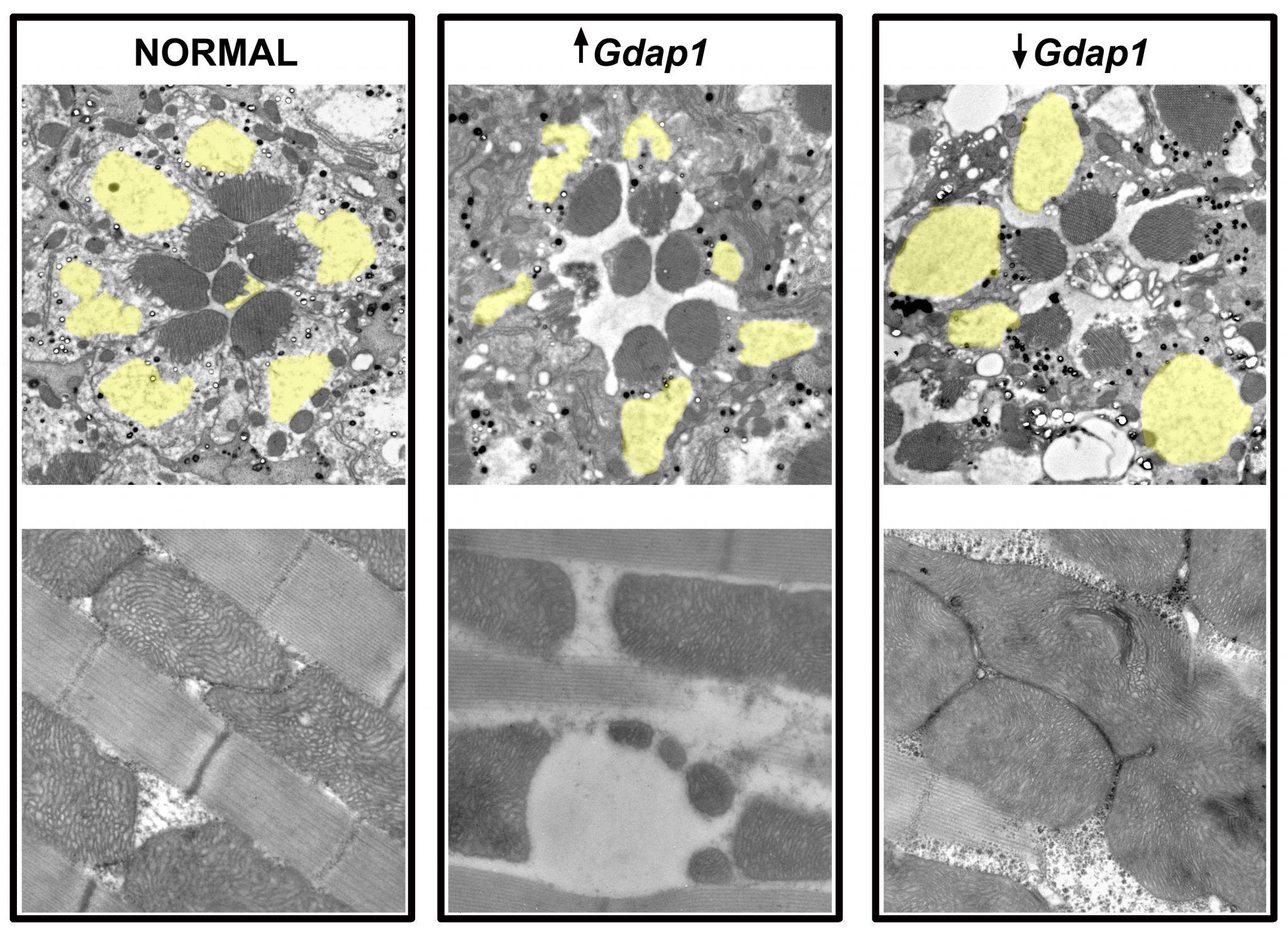
Primero, los investigadores han validado que Drosophila contiene el gen ortólogo al GDAP1 humano, al cual han nombrado como Gdap1. Además, han estudiado la localización de Gdap1 confirmando que se ubica en la mitocondria y que está relacionada con los procesos de fisión y fusión mitocondrial.
La generación del modelo engloba tres aproximaciones: el estudio de los posibles defectos provocados por la sobreexpresión de Gdap1; la afectación que desencadena el silenciamiento de Gdap1 y comprobar si los defectos causados por dicho silenciamiento son rescatados con la sobreexpresión de GDAP1 humano, confirmando así la existencia de una homología funcional.
Con dicho modelo, los investigadores han demostrado que la sobreexpresión y silenciamiento de Gdap1 causan una pérdida neuronal que se incrementa con la edad; los niveles de Gdap1 modulan la morfología mitocondrial y que tiene un papel importante para el correcto posicionamiento de la mitocondria en las neuronas.
Por otra parte, en el modelo de Drosophila, demuestran que Gdap1 tiene un papel importante en la integridad del músculo, ya que tanto la sobreexpresión como el silenciamiento de Gdap1 provocan una pérdida de los músculos encargados del vuelo de la mosca.
Por último, los estudios metabólicos concluyen que el daño primario es mitocondrial y el estrés oxidativo jugaría un papel más importante con la edad.
De esta manera, los investigadores ponen de manifiesto que Drosophila es un buen modelo para estudiar y entender las neuropatías axonales provocadas por mutaciones en GDAP1, y otros genes relacionados con la dinámica mitocondrial. El modelo puede ser útil para estudiar las diferentes mutaciones clínicas, buscar biomarcadores y desarrollar nuevos tratamientos.
Referencia: López del Amo V, et al. Mitochondrial defects and neuromuscular degeneration caused by altered expression of Drosophila Gdap1: implications for the Charcot–Marie–Tooth neuropathy. Hum Mol Genet. 2014 Aug 13. pii: ddu416.
Afiliaciones:
Programa de Enfermedades Genéticas Raras. Centro de Investigación Príncipe Felipe, Valencia. España: Víctor López del Amo, Máximo Ibo Galindo
Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER). España: Víctor López del Amo, Marta Seco-Cervera, José Luís García-Giménez, Federico V. Pallardó, Máximo Ibo Galindo
Departamento de Fisiología, Facultad de Medicina, Universitat de València, Instituto de Investigación Biomédica INCLIVA. Valencia, España: Marta Seco-Cervera, José Luís García-Giménez, Federico V. Pallardó
Department of Biomedical Science, University of Sheffield, Sheffield. Reino Unido: Alexander J. Whitworth