
José F. Moruno-Manchón, María P. Blasco-Conesa y Andrey S. Tsvetkov
Dept. Neurobiología y Anatomía, University of Texas McGovern Medical School, Houston, TX
La enfermedad de Huntington (EH) se caracteriza por movimientos involuntarios, cambios de humor, y demencia. Es una enfermedad que actualmente no tiene cura y los pacientes mueren 10-20 años después de la aparición de los primeros síntomas. La EH es causada por expansiones de poli-glutamina en la proteína huntingtina (Htt). Esta proteína mutante causa la degeneración de neuronas particularmente del cuerpo estriado, además del córtex y el hipocampo (Finkbeiner, 2011).
Durante la última década, las terapias para reducir la neurodegeneración de los pacientes de la EH se han enfocado en regular la actividad de las histonas deacetilasas (HDACs). Los inhibidores farmacológicos de las HDACs promueven la supervivencia en diferentes modelos de neurodegeneración. La actividad de HDAC1/2 puede ser inhibida por el fosfolípido esfingosina-1-fosfato (S1P) (Hait et al, 2009). S1P es un segundo mensajero implicado en multitud de vías de señalización, tanto a nivel extracelular, citoplasmático como nuclear. S1P es sintetizado a partir de la esfingosina a través de la fosforilación por las esfingosinas quinasas 1 (SK1) y 2 (SK2). En células proliferativas, SK1 se encuentra en el citoplasma, mientras que SK2 es mayoritariamente nuclear e incluso mitocondrial. SK1 está implicada en procesos relacionados con la supervivencia y la proliferación celular. Sin embargo, el papel de SK2 parece ser más complejo y poco se conoce sobre su función en neuronas.
En el laboratorio del Dr. Andrey Tsvetkov, en la Universidad de Texas en Houston, trabajamos con un modelo neuronal obtenido a partir de embriones de rata. El córtex y el cuerpo estriado del cerebro de los embriones de rata son diseccionados y homogeneizados, y la suspensión celular se siembra en placas de plástico. En el laboratorio utilizamos un novedoso método para cuantificar la supervivencia neuronal. Las neuronas son transfectadas con una proteína roja fluorescente, mApple, para visualizar la morfología neuronal. Gracias a un microscopio automatizado podemos seguir el mismo grupo de neuronas a lo largo del tiempo y determinar cómo las neuronas sobreviven a los diferentes tratamientos (Tsvetkov AS et al., 2013).
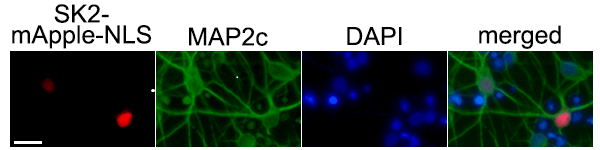
Por immunocitoquímica, comprobamos que SK2 se localiza principalmente en el núcleo neuronal. Para estudiar el papel de SK2 en el núcleo, diseñamos un ADN plasmídico que contiene la secuencia de la quinasa SK2 fusionada con una proteína roja fluorescente, mApple, y una secuencia de localización nuclear (NLS). Las neuronas que expresan SK2-mApple-NLS tienen un mayor riesgo de mortalidad, indicando que SK2 es tóxico en las neuronas primarias. La expresión de SK2-mApple-NLS en neuronas supone un incremento de siete veces la concentración de S1P en el núcleo, comparado con las muestras control. En células cancerosas, S1P se une a HDAC1/2 e inhibe su función deacetilasa (Hait et al., 2009), desencadenando la rotura de la doble cadena de ADN, la lesión del genoma más letal. Si esta lesión no es reparada por la maquinaria de reparación del ADN, el genoma acumula errores y la célula sufre inestabilidad genómica. Nos planteamos que SK2 podría inducir la muerte celular a través del daño del ADN. Efectivamente, la expresión de SK2-mApple-NLS induce el daño del genoma neuronal. Además, la expresión de SK2-mApple-NLS induce la acumulación de acetilación de la histona H4, indicando que SK2 inhibe la actividad de HDAC1/2. Estos efectos son reducidos por un inhibidor específico de SK2, ABC294640.
Creemos que SK2 puede formar parte de un mecanismo patológico de neurodegeneración. Utilizamos un modelo de ratón que reproduce muchas características observadas en pacientes de la EH, BACHD (Gray M et al., 2008). En el cerebro de los ratones BACHD adultos encontramos que existe un alto grado de lesión del DNA. Además, SK2 está hiperfosforilado en el córtex y en el cuerpo estriado de los ratones BACHD comparado con los ratones wild-type. Estos datos sugieren que la actividad quinasa de SK2 puede jugar un papel importante en el desarrollo de la EH.
A continuación, quisimos comprobar si la inhibición de la actividad quinasa de SK2 podría reducir la neurotoxicidad inducida por la proteína mutante Htt. Para ello utilizamos dos modelos neuronales de la EH: neuronas primarias de embriones de rata son transfectadas con un plásmido que contiene el exón 1 de la proteína Htt con una extensión de 72 glutaminas; y neuronas obtenidas a partir del córtex y del cuerpo estriado de embriones de ratones BACHD. El tratamiento con ABC294640 y la supresión de la expresión de SK2 con siRNA reducen el riesgo de mortalidad neuronal inducido por la expresión de la proteína mutante Htt.
En resumen, SK2 se expresa en el núcleo de neuronas primarias, SK2 nuclear es tóxico para las neuronas del córtex y del cuerpo estriado, SK2 induce daño del ADN e inhibe la actividad de las HDACs, SK2 esta hiperactivado en el cerebro de ratones BACHD, y la inhibición de la actividad de SK2 reduce la toxicidad inducida por la expresión de la proteína mutante Htt. Estos datos nos han permitido identificar una posible diana terapéutica para desarrollar nuevos tratamientos para la EH.
Referencia: Moruno-Manchon JF, et al. Inhibiting sphingosine kinase 2 mitigates mutant huntingtin-induced neurodegeneration in neuron models of Huntington disease. Hum Mol Genet. 2017 Feb 8. doi: http://dx.doi.org/10.1093/hmg/ddx046
Bibliografía:
Finkbeiner S. Huntington’s Disease. Cold Spring Harb Perspect Biol. 2011 Jun 1;3(6). pii: a007476. doi: 10.1101/cshperspect.a007476.
Gray M, et al. Full-length human mutant huntingtin with a stable polyglutamine repeat can elicit progressive and selective neuropathogenesis in BACHD mice. J Neurosci. 2008 Jun 11;28(24):6182-95. doi: 10.1523/JNEUROSCI.0857-08.2008.
Hait NC, et al. Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science. 2009 Sep 4;325(5945):1254-7. doi: 10.1126/science.1176709.
Tsvetkov AS, et al. Longitudinal imaging and analysis of neurons expressing polyglutamine-expanded proteins. Methods Mol Biol. 2013;1017:1-20. doi: 10.1007/978-1-62703-438-8_1.

