Un equipo de investigadores del Hospital Infantil de Filadelfia ha logrado aplicar por primera vez una terapia CRISPR personalizada en un paciente humano. El destinatario de este tratamiento pionero es KJ, un bebé con una enfermedad metabólica rara y potencialmente mortal, que ha mostrado signos de mejoría tras recibir la terapia, diseñada específicamente para corregir su mutación genética.
El tratamiento, publicado en The New England Journal of Medicine, supone un importante avance en el campo de edición genética aplicada a enfermedades monogénicas graves. Para modificar el genoma del paciente los investigadores han recurrido a los editores de bases, segunda generación de herramientas CRISPR, capaces de corregir directamente mutaciones sin cortar la doble hélice del ADN. Aunque todavía se encuentra en una fase experimental, el tratamiento sienta un precedente para el desarrollo de terapias génicas personalizadas, especialmente en enfermedades raras con opciones terapéuticas limitadas.
“Años y años de avances en edición genética y colaboración entre investigadores y clínicos han hecho posible este momento, y aunque KJ es sólo un paciente, esperamos que sea el primero de muchos que se beneficien de una metodología que puede adaptarse a las necesidades de cada paciente”, ha destacado Rebecca Ahrens-Nicklas, directora del Programa de Terapia Génica para Trastornos Metabólicos Hereditarios del Hospital Infantil de Filadelfia, profesora adjunta de Pediatría en la Facultad de Medicina Perelman de la Universidad de Pensilvania y una de los directores del trabajo.

Un diagnóstico rápido que abrió una vía terapéutica
El proceso que ha llevado desde el diagnóstico hasta la administración de la terapia CRISPR personalizada a KJ comenzó poco después de su nacimiento. Con apenas dos días de edad, un médico del hospital notó que algo no iba bien. KJ estaba letárgico y no comía bien.
Un análisis de sangre reveló niveles extremadamente altos de amonio en la sangre de KJ, lo que apuntaba a un trastorno metabólico. Al realizar más pruebas, el pequeño fue diagnosticado con deficiencia grave de carbamoil fosfato sintetasa 1 (CPS1), una enfermedad congénita muy poco frecuente que afecta a 1 de cada 1.300.000 recién nacidos. La deficiencia de CPS1 está causada por mutaciones en las dos copias del gen CPS1 que codifica una enzima clave en el ciclo de la urea, un proceso hepático necesario para eliminar el amonio, subproducto tóxico del metabolismo de las proteínas.
En los niños afectados, como ocurría con JK, la acumulación de amonio puede desencadenar síntomas graves en pocos días: vómitos, hipotonía, convulsiones y coma. Sin tratamiento adecuado, la enfermedad puede causar daños irreversibles o incluso la muerte.
Las medidas terapéuticas convencionales para la deficiencia en CPS1 incluyen una dieta restringida en proteínas, medicamentos para eliminar el nitrógeno y, en los casos más graves, como el de KJ, trasplante hepático. Sin embargo, para KJ la gravedad de su enfermedad imposibilitaba el trasplante como opción inmediata. Esta situación impulsó al equipo médico a buscar una alternativa experimental que pudiera estabilizar su estado clínico y ofrecer una esperanza de tratamiento a largo plazo.
Afortunadamente, antes de que KJ naciera, un equipo de científicos del Hospital Infantil de Filadelfia y de la Universidad de Pensilvania (Penn) ya estaba investigando cómo utilizar la edición genética para desarrollar tratamientos personalizados para trastornos metabólicos genéticos poco frecuentes. Tras conocer el caso de KJ, Rebecca Ahrens-Nicklas, pediatra genetista y directora del programa de Terapia Génica para Enfermedades Metabólicas Hereditarias y Kiran Musunuru, cardiólogo genetista de la Universidad de Pennsylvania, plantearon a la familia la posibilidad de participar en un ensayo clínico.
Tras evaluar sus opciones, la familia aceptó. “Pensamos que era nuestra responsabilidad ayudar a nuestro hijo, así que cuando los médicos acudieron a nosotros con su idea, depositamos nuestra confianza en ellos con la esperanza de que pudiera ayudar no sólo a KJ, sino a otras familias en nuestra situación”, ha señalado la la madre de KJ, Nicole Muldoon.
De la secuenciación genética a una terapia CRISPR personalizada en apenas unos meses
Una vez diagnosticado, el proceso que llevaría a una terapia personalizada basada en CRISPR fue rápido. En este proceso participaron tanto investigadores del Hospital Infantil de Filadelfia y la Universidad de Pensilvania, como diversas empresas de biotecnología.
Tras identificar las dos mutaciones responsables de la enfermedad —una heredada de cada progenitor— el primer paso fue seleccionar una de ellas como objetivo para llevar a cabo la corrección mediante edición genética. A continuación los investigadores diseñaron un editor de bases capaz de corregir la mutación puntual.
En el campo de la edición terapéutica del genoma, los editores de bases ya habían sido utilizados con éxito para curar una leucemia linfoblástica grave y sin tratamiento en diferentes pacientes pediátricos. La diferencia es que en estos casos no se trataba de tratamientos personalizados. Además, se trataba de terapia ex vivo, donde le modificaban células extraídas de los pacientes, que eran introducidas de nuevo tras la modificación. También se ha utilizado en una estrategia para regular los niveles de colesterol basada en la inactivación de la expresión del gen PCSK9. Más recientemente se está investigando su utilización en enfermedades como la distrofia de Duchenne.
La herramienta de edición fue validada en modelos celulares derivados del paciente y posteriormente en ratones humanizados. Para evitar una posible reacción inmunitaria adversa, los investigadores empaquetaron las instrucciones necesarias para producir los componentes de la terapia en nanopartículas lipídicas dirigidas al hígado (principal tejido donde se produce la proteína CPS1), similares a las utilizadas en algunas vacunas de ARN mensajero.
Una vez superadas las pruebas preclínicas y obtenida la aprobación regulatoria de la FDA para un uso compasivo (utilización cuando una enfermedad es grave y no hay alternativas terapéuticas), se inició el tratamiento tan solo siete meses tras el diagnóstico.
En qué consisten la terapia CRISPR personalizada de KJ y cómo actúan los editores de bases
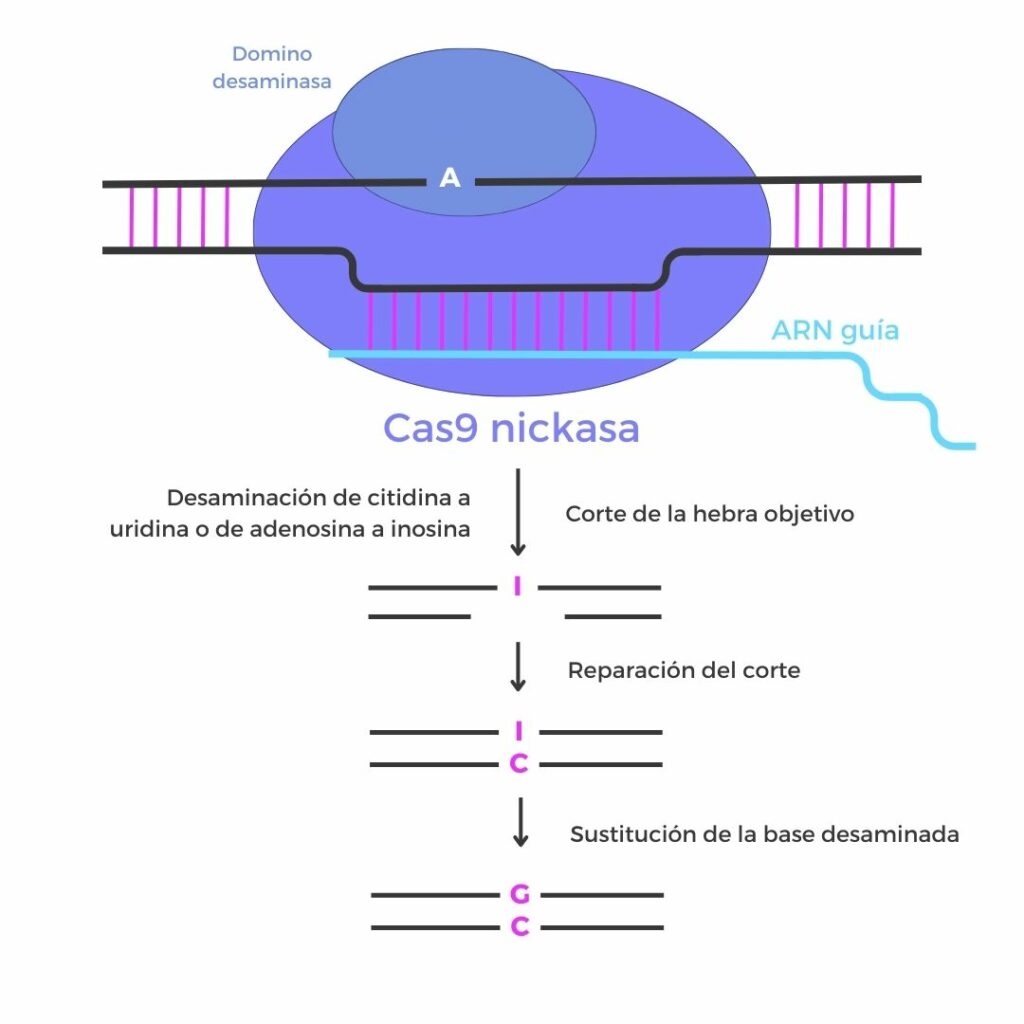
La terapia administrada a KJ se basa en una técnica avanzada de edición genética conocida como edición de bases, basada en las herramientas CRISPR. Los editores de bases permiten realizar cambios precisos en el ADN sin necesidad de cortar ambas cadenas de la doble hélice, como hacen las herramientas CRISPR de primera generación. Esta aproximación reduce el riesgo de inserciones o deleciones indeseadas y, por tanto, de efectos fuera del ADN que se quiere modificar.
El mecanismo de acción de los editores de bases, desarrollado por David Liu, investigador del instituto BROAD, es sencillo. El sistema CRISPR tradicional consta de una enzima Cas9 que corta el ADN y un ARN guía que posiciona a Cas9 en el lugar del ADN que se quiere editar. Los editores de bases utilizan una proteína Cas9 inactiva, fusionada a una enzima que promueve un cambio concreto de base (o letra) en el ADN. Para curar la enfermedad de JK, los investigadores desarrollaron un editor de bases tipo ABE (Adenine Base Editor), diseñado para convertir una adenina (A) en una guanina (G) en una posición determinada del gen CPS1. Con esta modificación personalizada pretendían restaurar parcialmente la función de la CPS1 en el hígado.
Las instrucciones de los editores de bases fueron empaquetadas en nanopartículas lipídicas similares a las utilizadas en algunas vacunas de ARN mensajero, para facilitar la entrada del material genético en las células hepáticas sin generar una respuesta inmunitaria significativa. Una vez en estas células, los componentes del sistema de edición se encargarían de modificar el ADN.
Otra ventaja de la terapia es que está diseñada para actuar únicamente sobre células somáticas, de modo que cualquier modificación genética permanece limitada al paciente tratado y no se transmite a la descendencia.
Resultados preliminares: mejoras clínicas sin efectos adversos graves
Desde la primera administración, en febrero de 2025, KJ ha mostrado signos consistentes de mejora. En primer lugar, ha podido aumentar la ingesta de proteínas sin que se produzca una elevación de los niveles de amonio en sangre. Además se ha reducido la necesidad de medicación y ha superado episodios de infecciones virales sin complicaciones metabólicas.
Según los investigadores, hasta abril de 2025 no se han observado efectos adversos graves relacionados con la terapia y las perspectivas son son prometedoras. Aunque ha pasado poco tiempo y todavía se desconoce la proporción de células editadas en el hígado de KJ, ya que no se ha realizado biopsia hepática (por ser una intervención invasiva no recomendada en lactantes), los marcadores clínicos y funcionales indican que la corrección genética ha tenido un efecto terapéutico significativo.
En cualquier caso, el niño será monitorizado durante años para evaluar tanto la duración de la respuesta como la seguridad a largo plazo. Este seguimiento también será necesario para descartar que se hayan producido cambios no deseados en el ADN o determinar si serán necesarias nuevas dosis de terapia.
Terapia CRISPR personalizada: potencial transformador y límites actuales
El caso de KJ es una prueba de concepto del potencial de las herramientas CRISPR para el desarrollo de tratamientos personalizados en pacientes con enfermedades muy poco frecuentes.
Expertos como Lluís Montoliu o Gemma Marfany han destacado a Science Media Center el valor científico del procedimiento y su rigor en todas las fases del desarrollo. “Realmente es un caso único, una prueba de concepto con éxito, diseñada y aplicada en un tiempo récord, en el que los investigadores y clínicos no se han saltado ni uno de los pasos previos preclínicos, ya que han generado modelos celulares humanos y también un modelo de ratón humanizado con la mutación del paciente para comprobar la seguridad de la dosis y la eficiencia de la estrategia terapéutica”, ha señalado Gemma Marfany. “Además, han contado con todas las aprobaciones de los comités de bioética correspondientes. Me parece un ‘milagro’ científico que ha permitido curar una enfermedad severa muy minoritaria, y aporta conocimiento para tratar muchas otras enfermedades”.
Sin embargo, ambos expertos también señalan sus limitaciones: el alto coste, la dificultad de replicar estos tratamientos en entornos clínicos más amplios y la incertidumbre sobre la equidad y accesibilidad en el futuro. “Desde el punto de vista experimental, el abordaje de terapias de un solo paciente es muy arriesgado, dado que la falta de controles o de variables hace que solamente se pueda explicar el éxito del tratamiento, cuando ocurre, pero no el fracaso de este, que puede estar causado por múltiples factores que no se han controlado en el diseño experimental”, ha señalado Lluís Montoliu. “La FDA permite este tipo de tratamientos experimentales en casos de enfermedades muy graves, como es la deficiencia en CPS1, incurables y con un elevado peligro de fallecimiento”.
Algunas cuestiones sobre los ensayos o terapias para un único paciente se plantearon hace unos años tras el desarrollo del fármaco milasen, diseñado de forma personalizada para una niña con enfermedad de Batten. El milasen, que consistía en unos ácidos nucleicos diseñados para regular la expresión de un gen (y no implicaba modificación del ADN), se diseñó, desarrolló y aprobó en menos de dos años, entre 2016 y 2018. El ensayo clínico personalizado para KJ, iniciado en apenas 7 meses tras el diagnóstico, muestra cómo se ha acortado el tiempo de desarrollo de este tipo de tratamientos, con los recursos e infraestructura adecuados.
En cualquier caso, la terapia personalizada con editores de bases plantea un futuro muy prometedor para la medicina de precisión para enfermedades monogénicas. La combinación de editores de bases y sistemas de administración podría permitir el tratamiento de un número creciente de enfermedades raras graves, a medida que mejoran la eficiencia, la seguridad y la escalabilidad de estas tecnologías.
“Queremos que todos y cada uno de los pacientes tengan la posibilidad de experimentar los mismos resultados que vimos en este primer paciente”, ha señalado Kiran Musunuru. “La promesa de la terapia génica de la que hemos oído hablar durante décadas se está haciendo realidad, y va a transformar por completo la forma en que abordamos la medicina”.
Artículo científico:
Musunuru K, et al. Patient-Specific In Vivo Gene Editing to Treat a Rare Genetic Disease. N Engl J Med. 2025 May 15. doi: https://doi.org/10.1056/nejmoa2504747
Otras fuentes:
The Future of Personalized Medicine is Here: KJ’s Story. https://www.chop.edu/centers-programs/genetherapy4inheritedmetabolicdisorders/future-personalized-medicine-here-kjs
World’s First Patient Treated with Personalized CRISPR Gene Editing Therapy at Children’s Hospital of Philadelphia. https://www.chop.edu/news/worlds-first-patient-treated-personalized-crispr-gene-editing-therapy-childrens-hospital
CORRECCIÓN 02-06-2025: Se corrige la frecuencia de la deficiencia en CPS1, que afecta a 1 de cada 1.300.000 recién nacidos y no 1 de cada 300.000 como se había indicado inicialmente.




