- Investigadores del CNIC, liderados por el Dr. José Jalife, han hecho un descubrimiento clave sobre las arritmias cardíacas en el síndrome de Andersen-Tawil (ATS).
- La mutación Kir2.1 C122Y está vinculada directamente a estas arritmias mortales, según un estudio publicado en Circulation Research.
- Este hallazgo podría abrir la puerta a tratamientos personalizados, marca un hito en la comprensión de las arritmias cardíacas y ofrece esperanza para prevenir la muerte súbita en diversas enfermedades cardíacas.
Un equipo del Centro Nacional de Investigaciones Cardiovasculares (CNIC) ha hecho un descubrimiento trascendental en la comprensión de las arritmias cardíacas, al desvelar los misterios del síndrome de Andersen-Tawil (ATS), un trastorno cardíaco hereditario extremadamente raro. Liderado por el Dr. José Jalife, jefe de Grupo de Arritmias Cardíacas del CNIC, el estudio demuestra que la mutación genética especifica (C122Y) en un canal de potasio (Kir2.1) no solo altera la función de ese canal sino también la del canal principal de sodio (NaV1.5) del corazón, estableciendo un vínculo directo con las arritmias potencialmente letales asociadas al síndrome de Andersen-Tawil 1 (ATS1).

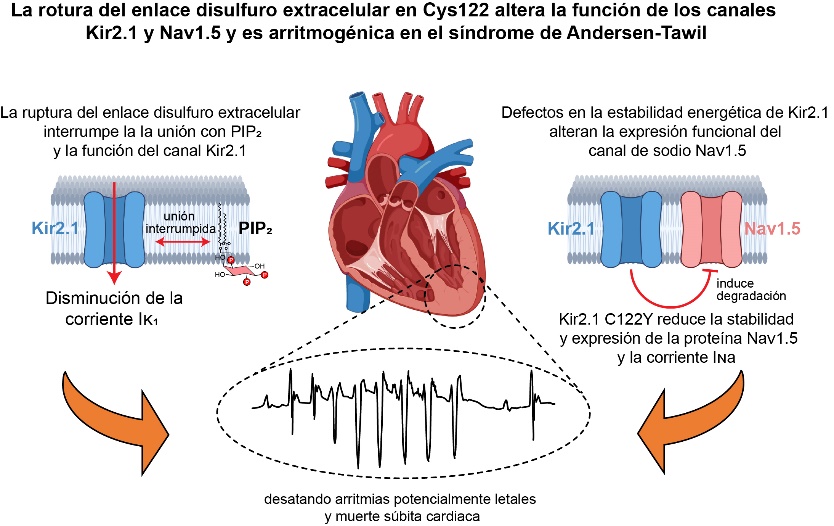
El estudio, publicado en la revista Circulation Research, revela por primera vez que la mutación C122Y en el canal Kir2.1 tiene un efecto doble sobre los canales Kir2.1 y NaV1.5: conduce a una reorganización del canal Kir2.1 que desestabiliza su unión con el fosfolípido PIP2, regulador principal en la membrana celular, y también desestabiliza la expresión de la proteína que forma el canal NaV1.5, reduciendo su función. La interacción de estos dos canales es vital para garantizar un ritmo cardíaco estable.
Se calcula que una de cada 3 personas padecerá una arritmia cardiaca a lo largo de su vida, aunque el síndrome de Andersen-Tawil afecta a menos de 1 por cada millón de personas. Está caracterizado por una tríada de síntomas que incluyen parálisis periódica, arritmias cardíacas y rasgos faciales distintivos. Las mutaciones en el gen KCNJ2, responsable del canal Kir2.1, heredadas de manera autosómica dominante, son las causantes del ATS tipo 1.
Los investigadores han ido más allá, creando un modelo de ratón que replica las principales irregularidades eléctricas cardíacas en pacientes con ATS1. Este modelo reveló sorprendentemente que la mutación no solo afecta a Kir2.1, sino que también interfiere con la estabilidad y expresión del canal NaV1.5 en el corazón, reduciendo su función.
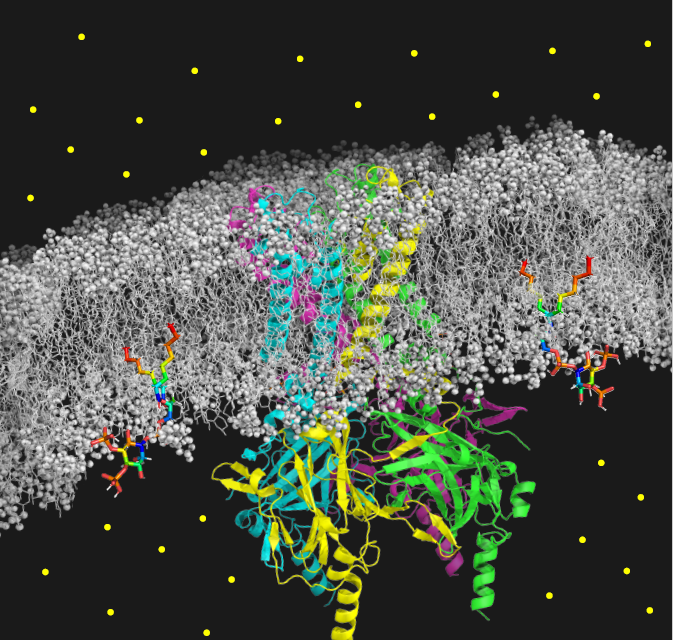
El Dr. Jalife subraya la relevancia de esta interacción para la salud cardíaca, advirtiendo que cualquier alteración que perturbe la función de uno o de ambos canales puede desatar arritmias potencialmente mortales.
“El descubrimiento de que una mutación considerada ‘monogénica’ no solo perturba al canal iónico afectado directamente por la mutación, sino también la función de un canal complementario, ambos fundamentales para la excitabilidad y la conducción eléctrica del corazón, supone una mayor comprensión de los mecanismos asociados a las arritmias cardíacas asociadas no solo con ATS1, sino también con patologías cardiacas más habituales”, señala el Dr. Francisco Cruz, responsable principal del estudio.
Y añade que los resultados “respaldan la hipótesis de que los mecanismos moleculares varían según la mutación específica, abriendo la puerta a tratamientos farmacológicos y manejo clínico personalizados para cada paciente”.
Este avance, concluyen los investigadores, representa un hito en la comprensión y abordaje de las arritmias cardíacas, ofreciendo un paradigma revolucionario con potencial impacto clínico. Mejorar el conocimiento sobre la función del canal Kir2.1 allana el camino para enfoques innovadores y seguros que podrían prevenir la muerte súbita en diversas enfermedades cardíacas, beneficiando a millones de personas en todo el mundo.
Además, sentará las bases para la generación de enfoques innovadores, efectivos y seguros para prevenir la muerte súbita en estas y otras enfermedades cardíacas devastadoras. En comparación con datos anteriores, todos los resultados presentados aquí respaldan la hipótesis de que los mecanismos moleculares que aumentan la susceptibilidad a las arritmias y la muerte súbita cardíaca en ATS1 son diferentes dependiendo de la mutación específica, por lo que el tratamiento farmacológico y el manejo clínico deberían ser diferentes para cada paciente.
Este estudio ha sido financiado por el Instituto Nacional del Corazón, los Pulmones y la Sangre de los NIH USA; la Fundación ‘la Caixa; Fundación La Marató de TV3; CIBERCV; Programa Horizon 2020 de la Unión Europea, y Programa S2022/BMD7229. Los estudios de imagen se realizaron en el nodo TRIMA@CNIC de la ICTS ReDIB.
Fuente: Cruz FM, et al. Extracellular Kir2.1C122Y Mutant Upsets Kir2.1-PIP2 Bonds and Is Arrhythmogenic in Andersen-Tawil Syndrome. Circ Res. 2024 Apr 12;134(8):e52-e71. doi: 10.1161/CIRCRESAHA.123.323895.
Si te ha gustado esta noticia y quieres aprender más sobre Genética en Medicina, te interesan nuestros cursos y formación universitaria, como el “Experto en Cardiogenética”.



